امراض الدم الوراثية بالتفصيل و بالعربي
صفحة 1 من اصل 1

 امراض الدم الوراثية بالتفصيل و بالعربي
امراض الدم الوراثية بالتفصيل و بالعربي
من طرف DREAM MAN الخميس يناير 27, 2011 11:47 pm
السلام عليكم و رحمة الله و بركاته ,,,,
أمراض الدم الوراثية ,,
الثلاسيميــــــــــــــــا :
الثلاسيميا من أهم أمراض الدم الوراثية الانحلالية- التي تسبب تكسر كريات
الدم الحمراء- الشائعة على مستوى العالم بشكل عام وعلى مستوى منطقة البحر
الأبيض المتوسط بشكل خاص ، و الثلاسيميا نوعان الألفا و البيتا وحين يأتي
الحديث عن هذا المرض فإننا نقصد به (البيتا ثلاسيميا ).
و الثلاسيميا كلمه يونانية الأصل تعني فقر دم منطقة البحر الأبيض المتوسط
حيث أن هذا المرض عرف واشتهر في هذه المنطقة بشكل كبير و ، ويعرف أيضا باسم
أنيميا البحر المتوسط (Mediterranean anemia) وفي الولايات المتحدة
الأمريكية كان يعرف باسم أنيميا كوليز (Cooleys anemia).وفي منطقة البحر
الأبيض المتوسط كان لا بد أن يخضع كل مولود جديد بعد ولادته بشهور لهذا
الفحص لمعرفة ما إذا كان حاملا أو مصابا بالبيتا ثلاسيميا .أما في منطقة
الخليج العربي يحمل كل فرد من بين 20 فرد لهذا المرض أي نسبه حاملي صفة
المرض في الخليج 5 % وهي نسبه لا يمكن تجاهلها حيث أنه في كل مرة تحمل فيه
امرأة حاملة للمرض ومتزوجة من رجل أيضا حامل للمرض فإن احتمال إصابة
الجنين تصل إلى 25% وان اكثر من 60% من أطفال هذه الأسرة السليمين أيضاً
حاملين للمرض. وتكمن مشكلة المرض في عدم قدرة الجسم من تكوين كريات الدم
الحمراء - التي تنقل الغذاء والأكسجين إلى مختلف أنحاء الجسم – بشكل سليم
نتيجة لخلل في تكوين الهيموجلوبين( خضاب الدم) أدى إلا عدم اكتمال نضج
الكريه الحمراء و تكسرها وتحللها بعد فتره قصيرة من إنتاجها (يذكر أن عمر
كريه الدم الحمراء تكون 120 يوما) لذ فان المريض يحتاج إلى نقل دم بشكل
دوري كل 3-4 أسابيع حسب عمره و درجه نقص الهيموجلوبين .
ينتشر المرض في عدة مناطق من العالم ولكنها تكثر في المناطق التالية:
* حوض البحر الأبيض المتوسط ( اليونان، مالطا، قبرص، تركيا و إيطاليا).
* منطقة الخليج العربي.
* منطقة الشرق الأوسط وتشمل إيران العراق سوريا الأردن وفلسطين.
*شمال أفريقيا وتشمل مصر تونس الجزائر المغرب وبعض من الدول الأفريقية الأخرى.
* جنوب شرق أسيا وتشمل تايلاند، الفلبين، إندونيسيا، سنغافورة، كمبوديا، فيتنام وماليزيا.
* شبه القارة الهندية.
* دول أخرى كالصين، أرمينيا، جورجيا، أذربيجان.
المورث (الجين) المسؤول عن إنتاج مادة الألفا جلوبين: يسمى بمورث الألفا
جلوبين يوجد على الكروموسوم رقم 16 ،ويوجد منه أربع نسخ (أي أربع جينات او
مورثات)اثنتان منها موجودة على كروموسوم رقم 16 الأتي من الأم والاثنان
الآخران موجودان على الكروموسوم الأتي من الأب. واي خلل في هذه المورثات قد
يسبب مشاكل صحية حسب عدد المورثات المعطوبة او المصابة بالطفرة، واليك بعض
التفصيل
الألفا ثلاسيميا الساكتة Silent alpha Thalassemia):
فإذا أصابت الطفرة مورث واحد فقط فأن الشخص لا يصاب بأي مشكلة صحية و تسمى
هذه الحالة بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia).وفي
العادة لا يعرف الشخص انه حامل لمورث معطوب وقد يصعب التعرف على هذه الحالة
حتى عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا
يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر
الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر إلا بالكشف
على المورثات مباشرة،وهذه غير متوفرة في الكثير من المراكز الطبية.
الحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait)
فلو أصاب العطب مورثين من هذه الأربعة فقط سمي الشخص حامل لصفة الألفا
ثلاسيميا وهو لا يصاب بأية مشكل صحية ولكن قد يولد له أطفال مصابون بمشاكل
صحية في الهيموجلوبين إذا كانت زوجته حاملة لنفس الصفة.
مرض بالهيموجلوبين اتش :
لو أصاب العطب(الطفرة)ثلاث مورثات من الأربعة يصاب الشخص بمرض يسمى
بالهيموجلوبين اتش (Hemoglobin H disease) .وعند ولادة الطفل المصاب بعطب
في ثلاثة مورثات من نوع ألفا يكون لدية قلة في كمية الهيموجلوبين في كريات
دمهم الحمراء مع وجود هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج
عن اتحاد أربع قطع من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة
التي تقل فيها كمية الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من
النوع المتوسط فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100
ملليتر ويكون لدية تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال
لنقل الدم، أما البالغين فقد يحتاجون لنقل كميات من الدم.
استسقاء شديد للجنين (Hydrops Fetalis):
لو أصاب العطب(الطفرة)جميع المورثات الأربعة يصاب الجنين وهو في بطن أمه
باستسقاء شديد في جميع الجسم نتيجة لوجود فقر دم حاد مع فشل في القلب
كنتيجة ثانوية لنقص الهيموجلوبين في الدم.
المورث (الجين) المسؤول عن إنتاج مادة البيتا جلوبين: يسمى بمورث البيتا جلوبين يوجد على الكروموسوم 11.
حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى)
تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب.
وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا
يعاني حامل المرض من أي مشكلة صحية إلا الهم من فقر دم طفيف ولا يحتاج إلى
نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك
نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من
السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد
صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك
إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا
شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا
حصيات في المرارة أو تقرح في ساق القدم.
حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى)
تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب.
وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا
يعاني حامل المرض من أي مشكلة صحية إلا الهم من فقر دم طفيف ولا يحتاج إلى
نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك
نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من
السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد
صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك
إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا
شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا
حصيات في المرارة أو تقرح في ساق القدم.
مصاب بالثلاسيميا المتوسط
تحدث الثلاسيميا المتوسطة عند وجود عطب(طفرة) في كلا مورثي البيتا جلوبين .
فينتج عن ذلك نقص متوسط الشدة في كمية البيتا جلوبين المنتجة في الجسم
وتؤدي في هذه الحالة إلى نقص متوسط الشدة لمستوى الهيموجلوبين في الدم
فيتراوح في العادة بين 7-10 غرام لكل 100 ملليمتر ،وفي العادة لا يحتاج
المريض لنقل دوري للدم ، لكنه مع التقدم في العمر و في خلال الحمل في حالة
المرأة قد يحتاج إلى نقل دم وأشراف طبي بشكل منتظم. و تشكل الثلاسيميا
المتوسطة من 2-10% من مرضى البيتا ثلاسيميا ،وتظهر أعراضها خلال ألسنه
الثانية إلى الرابعة من عمر المريض ، و مخبرياً تظهر علامات فقر الدم
خلال السنة الأولى من عمر المريض من خلال فحص الدم الروتيني.وقد يتعرض
المريض لبعض المشاكل الصحية كضعف البنية وتضخم الكبد والطحال و الإصابة
باليرقان(اصفرار الجلد والعينان).
مصاب بالثلاسيميا (الثلاسيميا الكبرى)
تحدث الثلاسيميا الكبرى أو الشديدة عند وجود عطب(طفرة) في كلا مورثي
البيتا جلوبين كما هو الحال في الثلاسيميا المتوسطة ولكن نوع العطب
(الطفرة)في مورث البيتا هذه المرة اشد فينتج عن ذلك نقص شديد نسبة البيتا
جلوبين فينقص بذلك الهيموجلوبين إلا نسب اقل من 7 غرام لكل 100 ملليليتر
نتيجة لتكسر كريات الدم الحمراء الغير طبيعية قبل انتهاء عمرها الافتراضي
(120 يوم). و يحتاج المريض لنقل دوري للدم كل 3-4 أسابيع للمحافظة على نسبه
عالية من الهيموجلوبين لكي ينموا الجسم بشكل صحي. ويمكن اكتشاف المرض خلال
الأشهر الستة الأولى من عمر الطفل
مريض بالثلاسيميا (الثلاسيميا الكبرى) + الأنيميا المنجلية
في هذه الحالة يصاب المريض بمرضين في أن واحد.فيكون الشخص أما لدية مرض
الأنيميا المنجلية ومرض الثلاسيميا معا أو يكون حاملا للأنيميا المنجلية مع
مرض الثلاسيميا.وقد تتراوح شدة المرض وانقص الهيموجلوبين على حسب شدة
الإصابة في مورث البيتا جلوبين.
الاعـــــــــراض :
تختلف أعراض المرض على حسب نوع الثلاسيميا المصاب بها الشخص.وقد لا يشعر
المصاب بالمرض بأي مشكلة كما هو الحال فالشخص الحامل للألفا ثلاسيميا
وتكتشف بالمصادفة عند إجراء تحليل دم لكريات الدم الحمراء والهيموجلوبين.و
في المقابل قد يكون شديد كما هو الحال في البيتا ثلاسيميا الكبر في تكون
الأعراض واضحة و تتفاقم المشكلة إذا لم ينقل للمريض دم بشكل دوري.كما أن
المرض قد يكون شديد لدرجة أن الجنين يموت في بطن أمة أو يولد مع استسقاء في
جميع الجسم نتيجة لنقص الهيموجلوبين في الدم وهبوط شديد في القلب،وهذا
يحدث في الألفا ثلاسيميا التي يكون فيها جميع مورثات(جينات)الألفا
هيموجلوبين معطوبة (بها طفرة مرضية).
ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال
وفيما يلي الأعراض التي تحدث للمصابين بمرض البيتا ثلاسيميا الكبرى إذا لم يعالج المريض:
.شحوب واصفرار البشرة والشفتين
الخمول و الشعور بالتعب والإرهاق لأقل جهد.
فقدان الشهية.
.نقص حاد في الهيموجلوبين (اقل من 9 مج\مل دم)
تضخم الكبد والطحال نتيجة عجز نخاع العظام عن إنتاج كريات الدم الحمراء
فيبدأ الكبد والطحال وأعضاء أخرى بمحاولة تصنيع الدم بنفسها مما يؤدي إلى
تضخمها.
الانعكاسات النفسية السيئة لدى المريض نتيجة شعوره بالمرض الدائم والنقص الصحي مقارنة بأقرانه.
التأخر في النمو الجسماني كالطول والوزن .
تغيرات في عظام الجسم ومنها الجمجمة ، بروز الجبهة وعظام الوجنتين ، انخفاض عظام الأنف وبروز عظام الفك العلوي.
زيادة الالتهابات بشكل عام.
سرعة ضربات القلب لمحاولة تعويض نقص الهيموجلوبين وذلك بزيادة سرعة ضخ القلب للدم، كذلك قد تحدث مضاعفات قلبيه مع مرور الوقت.
حدوث مضاعفات نتيجة تراكم الحديد في أعضاء مختلفة من الجسم أن لم ينتظم
المريض باستعمال الحقنه اليومية للدسفيرال والتي تؤدي إلى التخلص من الحديد
عن طريق البول وتتمثل هذه المضاعفات في تشحم الكبد ، اسوداد لون الجلد،
خلل هرموني (مرض السكري) ، انخفاض في هرمون الغدة الدرقية وهرمون الجار
درقية وهرمونات الغدة النخامية
مشاكل في القلب كتضخم عضلة القلب مع هبوط في القلب وعدم أنتظام في دقات القلب .
احتمال أصابه المريض بالأمراض المعدية نتيجة لنقل الدم بشكل مستمر و لكن
قلة تلك المخاطر بعد قيام جميع مراكز التبرع بالدم بفحص إذا ما كان دم
المتبرع بدم مصاب بالإيدز او فيروسات التهاب الكبد الوبائي.
الاسبـــــــاب :
الثلاسيميا مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض
أطفالهم.ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور
المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.ولكي تفهم هذا
الجانب العلمي أنصحك بالرجوع إلى صفحة أساسيات الوراثة لمعرفة ما هي
الخلية والنواة و الكروموسوم والمورث .وسوف أقوم بشرح مختصر عن هذا الكلمات
في سياق كلامي عن الهيموجلوبين والمورثات في هذه الصفحة.
تصنيع الهيموجلوبين:
يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع
العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها
البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم
(Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى
بالجلوبين(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع
بيتا وفي هذه الحالة يسمى هذا الهيموجلوبين بهيموجلوبين ( أ ) أو (A ).
هذا الترابط العجيب بين الحديد و الهيم و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم.
كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي
عبارة عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل
الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات(الصبيغات)
،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين
يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات
موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض
الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة)
في المورثات التي تصنع مادة الجلوبين.
المورثات و الجلوبين:
هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى
تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان
من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على
الكروموسومات،كما هو الحال في مادة الهيم.
ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم
11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11
الذي ورثة الإنسان من أمة..بينما يصنع مادة الألفا جلوبين من أربع
مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين
على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من
الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان
بمرض ألفا ثلاسيميا، و إذا أصاب الخلل مورث من مورثات البيتا يصاب الإنسان
بمرض البيتا ثلاسيميا.هذه بشكل مبسط العلاقة بين المورثات و
الثلاسيميا،ولكن الموضوع اعقد من ما ذكرنا ،ولكي تعرف المزيد تابع القراءة!
البيتا ثلاسيميا (Beta Thalassemia ) :
خلاصة ما ذكرنا أن هناك أربع مورثات لتصنيع مادة الألفا جلوبين، و مورثين
لتصنيع مادة البيتا جلوبين.واليك الآن بعض التفصيل في هذا الأمر و لنبدأ
بالبيتا ثلاسيميا.لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات
البيتا جلوبين فأن الخلية تستمر في إنتاج البيتا جلوبين وذلك نظرا لوجود
مورث أخر سليم .ولكن هناك نقص في الكمية الإجمالية المنتجة من هذه المادة
ولكن هذا النقص لا يسبب مرضا ولكن يسمى الشخص الذي يحمل مورث واحدا معطوبا
من مورثات البيتا بالحامل للمرض،أي انه حامل لمرض البيتا ثلاسيميا ويمكن
أن يتعرف على هذه الحالة بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)وهي
متوفرة في الكثير من المستشفيات.أما لو حدثت الطفرة في كلا النسختان فان
مجوع الكمية المصنعة من البيتا جلوبين تقل بشكل كبير فيصاب الشخص بمرض
البيتا ثلاسيميا وتكون شدة المرض على حسب شدة الطفرة فبعض الطفرات تسمح
بإنتاج كمية معقولة من مادة البيتا جلوبين(ويسمى المرض في هذه الحالة
بالبيتا ثلاسيميا الصغرى)وهناك طفرات شديدة تؤدي إلى نقص كبير وقد يكون
كامل في مادة البيتا جلوبين (ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا).
نظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض
على حسب عدد المورثات المعطوبة .فإذا أصيب مورث واحد فأن الشخص لا يصاب
بأي مشكلة صحية(يسمى بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia)
وقد يصعب التعرف على هذه الحالة عند إجراء تحليل دم عادي(أي عدد الكريات
الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة
الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه
يصعب اكتشاف هذا الأمر الا بالكشف على المورثات مباشرة،وهذه غير متوفرة في
الكثير ن المراكز الطبية.بينما فحص حركة الهيموجلوبين الإلكترونية (
Hemoglobin Electrophoresis) متوفر في الكثير من المستشفيات.
الالفا ثلاسيميا ( Alpha thalassemia):
بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم
16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم
16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة)
في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا.
ونظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض
على حسب عدد المورثات المعطوبة .فإذا أصابت الطفرة مورث واحد فقط فأن
الشخص لا يصاب بأي مشكلة صحية و تسمى هذه الحالة بالألفا ثلاسيميا الساكتة
Silent alpha Thalassemia).وفي العادة لا يعرف الشخص انه حامل لمورث معطوب
وقد يصعب التعرف على هذه الحالة حتى عند إجراء تحليل دم عادي(أي عدد
الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة
الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه
يصعب اكتشاف هذا الأمر إلا بالكشف على المورثات مباشرة،وهذه غير متوفرة في
الكثير من المراكز الطبية.أما إذا أصابت الطفرة مورثين اثنين من الأربعة
فان هذه الحالة تسمى بالحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia
Trait) وهذا الشخص أيضا لا يصب بمشاكل في الصحة وليس علية خطورة .ولكن
يمكن الاشتباه في أن الشخص لدية هذا النوع من الثلاسيميا عند إجراء تحليل
عادي لكريات الدم الحمراء ،ففي هذه الحالة تكون كريات الدم الحمراء اصغر من
الحجم المعتاد وكمية الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات
التي على كريات الدم الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال
المصابون بفقر الدم الناتج عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس
كمية الحديد في الدم فإذا كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن
الألفا ثلاسيميا .أما إذا كان هناك نقص في الحديد فيعالج الأمر بإعطاء
الحديد ثم يعاد التحليل .و إذا زال نقص الحديد واستمر صغر كريات الدم
الحمراء فان هذا يعني أن الشخص كان عنده نقص في الحديد مصحوب بألفا
ثلاسيميا.يكثر الاشتباه بين فقر الدم الناتج عن نقص الحديد و ألفا ثلاسيميا
لذلك على العائلة تنبيه الطبيب على أن بعض أفراد العائلة لديهم ألفا
ثلاسيميا لكي لا يعطى الطفل أو البالغ(كامرأة الحامل) جرعات من الحديد من
دون أن يكون هناك ضرورة لهذا الأمر لأن ارتفاع كمية الحديد في الجسم
مضر.أما إذا أصاب العطب(الطفرة)ثلاث مورثات من الأربعة فان الشخص يصاب بمرض
يسمى بمرض الهيموجلوبين اتش (Hemoglobin H disease).وعند ولادة الطفل
المصاب يكون هناك نقص في كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود
هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع
من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية
الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من النوع المتوسط
فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية
تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال لنقل الدم، أما
البالغين فقد يحتاجون لنقل كميات من الدم.أما إذا أصاب العطب(الطفرة)جميع
المورثات الأربعة يصاب الجنين باستسقاء شديد في جميع الجسم نتيجة لوجود فقر
دم حاد مع فشل في القلب كنتيجة ثانوية لنقص الهيموجلوبين في الدم.ويموت
الجنين إذا لم ينقل له جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل
دم بشكل دوري نتيجة لوجود فقر دم شديد إلى متوسط الشدة.
التشخيــــــــص :
الهيموجلوبين وفقر الدم الناتج عن تكسر الدم:
الهيموجلوبين عبارة عن بروتين موجود في كريات الدم الحمراء وظيفته نقل
الأكسجين من الرئة إلى كافة خلايا الجسم ،وهو يتكون من الهيم (مادة الحديد +
صبغه) و الجلوبين (البروتين). وهناك أنواع مختلفة من بروتينات الجلوبين
المهمة منها الألفا ،البيتا ، الدلتا و الجاما جلوبين .وتحدث المشكلة
عندما يحدث نقص في مكونات بروتين الجلوبين بسبب خلل جيني وكل زوج من
الجينات الآتية من الأم و الأب يعمل على تنظيم و ناتج بروتينات الجلوبين
في الجسم فمثلا إذا كان الجين المسؤول عن بروتينات البيتا جلوبين به خلل
فلن يكون ناتج التكوين كاملا وصحيحا مما يؤدي إلى خلل في تكوين سلاسل
البيتا وبالتالي يؤدي إلى مرض البيتا ثلاسيميا. كما انه اذا كان نفس الخلل
موجود في بروتين الألفا جلوبين فانه ينتج مرض الألفا ثلاسيميا. وكلا
الألفا و البيتا ثلاسيميا يعتبران من الأمراض الشائعة جدا في منطقة الشرق
الأوسط وبأنواع نادرة أيضاويتغير نوع الهيموجلوبين في الجسم خلال الحمل إلى
عدة شهور بعد الولادة نتيجة لتغير نوع الجلوبين المنتج:
فعند الولادة يكون اغلب الهيموجلوبين(بنسبة تزيد عن 80%) من ما يعرف باسم
الهيموجلوبين الجنيني ( Hb F) ويستمر تواجده إلى أن تتم عملية إنتاج
الهيموجلوبين البالغين( Hb A) بشكل منتظم خلال الستة شهور الأولى من
العمر.,كما يوجد نسبة قليلة (1.5-2.5%) من هيموجلوبين يسمى بالهيموجلوبين
البالغين نوع 2 (( Hb A2) ).
المعدل الطبيعي للهيموجلوبين عند البالغين الذكور = 13-16 جرام/ 100 ملليليتر وعند البالغات الإناث = 12-15 جرام/ 100ملليليتر .
حامل البيتا ثلاسيميا:
يصعب التاكد من ان الشخص حامل لثلاسيميا و لكن في العادة يكون مستوى
الهيموجلوبين طبيعي ولكن كريات الدم الحمراء اصغر من المعدل الطبيعي( MCV
اقل من 75).و عند اجراء فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin
Electrophoresis) يكون نسبة الهيموجلوبين A2 و هيموجلوبين F اعلى من
الطبيعي .
مصاب بالبيتا ثلاسيميا:
يكون مستوى هيموجلوبين في الدم اقل من 10 مليغرام لكل 100 مليلتر . و تكون
كريات الدم الحمراء اصغر من المعدل الطبيعي( MCV اقل من 75).و عند اجراء
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) يكون
نسبة الهيموجلوبين A2 و هيموجلوبين F اعلى من الطبيعي .
الألفا ثلاسيميا الساكتة Silent alpha Thalassemia):في العادة يصعب
التعرف على هذه الحالة فتحليل الدم طبيعي(أي عدد الكريات الحمراء
والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين
الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف
هذا الأمر إلا بالكشف على المورثات مباشرة (اي فحص DNA) ،وهذه غير متوفرة
في الكثير من المراكز الطبية.
حامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait) وهذا الشخص لا يصاب
بمشاكل في الصحة وليس علية خطورة .و لكن يمكن الاشتباه في أن الشخص لدية
هذا النوع من الثلاسيميا عند إجراء تحليل عادي لكريات الدم الحمراء ،ففي
هذه الحالة تكون كريات الدم الحمراء اصغر من الحجم المعتاد وكمية
الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات التي على كريات الدم
الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال المصابون بفقر الدم الناتج
عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس كمية الحديد في الدم فإذا
كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن الألفا ثلاسيميا .أما إذا
كان هناك نقص في الحديد فيعالج الأمر بإعطاء الحديد ثم يعاد التحليل .و
إذا زال نقص الحديد واستمر صغر كريات الدم الحمراء فان هذا يعني أن الشخص
كان عنده نقص في الحديد مصحوب بألفا ثلاسيميا.يكثر الاشتباه بين فقر الدم
الناتج عن نقص الحديد و ألفا ثلاسيميا لذلك على العائلة تنبيه الطبيب على
أن بعض أفراد العائلة لديهم ألفا ثلاسيميا لكي لا يعطى الطفل أو
البالغ(كامرأة الحامل) جرعات من الحديد من دون أن يكون هناك ضرورة لهذا
الأمر لأن ارتفاع كمية الحديد في الجسم مضر.قد يساعد تحليل حركة
الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) يكون نسبة
الهيموجلوبين A2 و هيموجلوبين F اعلى من الطبيعي .
مرض الهيموجلوبين اتش (Hemoglobin H disease) والذي يحدث نتيجة لوجود عطب
في ثلاث نسخ من الالفا جلوبين.عند ولادة الطفل المصاب يكون هناك نقص في
كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود هيموجلوبين غير طبيعي
يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع من جلوبين جاما .وبعد
مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية الجاما جلوبين في جسم
الإنسان) يصاب الطفل بفقر دم من النوع المتوسط فيتراوح مستوى الهيموجلوبين
حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان .
عطب أربع نسخ من مورثات الالفا جلوبين(استسقاء شديد للجنين): و يحدث
الاستسقاء نتيجة لوجود فقر دم حاد مع فشل في القلب كنتيجة ثانوية .مستوى
الهيموجلوبين منخفض.و في العادة يموت الجنين او يولد ميت إذا لم ينقل له
جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل دم بشكل دوري نتيجة لوجود
فقر دم شديد إلى متوسط الشدة. يمكن الكشف عن المرض باجراء حركة
الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) و يلاحظ عدم و جود
اي نسبة من هيموجلوبين A و لا A2 و لا F
العــــــــــــــلاج :
وسائل علاج البيتا ثلاسيميا:
نقل دم
ينقل الدم بشكل دوري للمريض (6-8 ملليليتر دم"كريات دم حمراء" لكل كجم من
وزن المريض كل 3-4 أسابيع)، ويجب ان يكون عمر الدم المنقول لا يتجاوز ال6
أيام. والنقل الدوري يحافظ على مستوى عالي من الهيموجلوبين لكي يصل
الأكسجين الى اجزاء الجسم بشكل افضل فيحافظ على النمو الطبيعي للطفل، ومنع
التغيرات التي تحدث في عظام الجسم،حماية القلب من مضاعفات فقر الدم، منع
تضخم الكبد والطحال.
ويحتاج المريض إلى كريات الدم الحمراء فقط لذا يجب ان يرشح الدم المنقول من الكريات البيضاء و الصفائح الدموية.
العلاج بالديسفسرال
يترسب الحديد في جسم المريض تدريجيا مما يؤدى إلى تعطيل وظائف الخلايا
وبالتالي إلى موتها وفقدان وظائفها، كما يؤدى إلى الكثير من اختلال الوظائف
الهرمونية و القلبية و الكبدية و الجلدية.
لذا يجب التخلص من الحديد الزائد (اكثر من 1000 مج\مل)
الديسفرال عبارة عن مادة ترتبط مع الحديد في الجسم وتخرج مرتبطة بالحديد
لخارج الجسم عن طريق البول، ومن هنا تأتى أهميه الديسفرال الذي يحقن عن
طريق الجلد لفتره8-10 ساعات يوميا او عن طريق الوريد،والعضل
إزالة الطحال
للطحال دور هام في التخلص من كريات الدم الميتة نتيجة مشاركة الطحال في
تعويض الجسم عن عجز نخاع العظم واستمرار تراكم الحديد بصوره كبيره يبدأ
بعدها الطحال بتدمير كريات الدم الحمراء والبيضاء والصفائح الدموية ،تتفاقم
حدة فقر الدم وتتناقص مناعة ومقاومة المريض ويصبح عرضة للنزف، ويتم
استئصال الطحال عندما تظهر المؤشرات التالية:
عندما يحتاج المريض إلى نقل كميات من الدم تزيد بمقدار واحد ونصف عن الكميه الكافية.
نقص حاد في كريات الدم البيضاء أو الصفائح الدموية نتيجة تدميرها في الطحال
التضخم الكبير والمتزايد للطحال.
زرع نخاع العظم
هذه العملية تعتمد على وجود متبرع يفضل أن يكون من أشقاء او شقيقات المريض
حيث يجب أجراء الفحوصات التي تؤكد من وجود التطابق النسيجي والخلوي (100%)
بين المتبرع والمريض،وتزداد فرص نجاح العملية للذين لا يعانون من ترسب
الحديد أو تضخم الكبد والطحال.
إن رفض الانسجه عادة ما يكون اقل نسبة في الأطفال عنه في الكبار فذلك لا
يحدث مباشرة بعد العملية إنما قد يحتاج إلى 3-5 سنوات يبقى فيها خطر
الرفض،فالمريض بحاجة إلى متابعة دقيقه للغاية.
ويتم فبل زرع نخاع العظام التخلص من نخاع المريض بتناوله الأدوية تدمر
خلايا النخاع وعلى مدى 6 أيام يشعر فيها بالوهن والضعف وتحت التعقيم التام
يتم شفط النخاع بواسطة حقنه خاصة وتحت التخدير العام وتستغرق هذه العملية
حوالي نصف الساعة وتستكمل العملية بإعادة حقن نخاع العظام السليم إلى
المريض فيما يشبه عملية نقل الدم فيجد النخاع الجديد تجويف نخاع المصاب.
والمتبرع عادة لا يعاني من أي مخاطر وجسمه قادر على تعويض الكميه التي تم
شفطها من نخاع العظام ولا يحتاج للبقاء في المستشفى اكثر من يوم واحد فقط.
الجديد في علاج البيتا ثلاسيميا:
1- بدأت بعض المراكز في إجراء عمليات نقل وزراعة نخاع العظم للجنين المصاب
داخل رحم أمه بدل إجهاضه وتتميز هذه العملية بندرة رفض جسم المريض للنخاع
المزروع.
2- يتوفر الان نوع مشابه لدواء الدسيفيرال الطارد للحديد من الجسم و يأخذ
عن طريق الفم بدل من الحقن التحت جلدية. و الابحاث المتتالية اثبتت فعالية
هذا الدواء مقارنة بالديسفيرال االذي يؤخذ عن طريق الحقن و يسمى هذا الدواء
الديفريبرو ن (deferiprone)و يسوق تحت اسم تجاري يدعى Ferriprox™.
طرق الوقاية:
فحوصات قبل الولادة
إذا كان كلا الوالدان يحملان صفة البيتا ثلاسيميا الصغرى فمن الممكن أن يتم
فحص الجنين للتأكد من سلامته من مرض الثلاسيميا خلال الأسبوع 10-11 من
الحمل. ويتم الفحص بأخذ عينه من المشيمة ( Chorionic Villus )وتحليلاها
بواسطة ال DNA لمعرفة إذا ما كان الجنين مصاب بالثلاسيميا أم لا.ولكنا يجب
أن يحضر لهذا الأمر فيجب أولا معرفة نوع الطفرة الموجودة في العائلة لكي
يستطيع الأطباء الكشف عنها خلال الحمل،وتظهر نتيجة التحليل خلال 3-4
أسابيع.وكما هو الحل في جميع الفحوصات الطبية فقد يكون هناك بعض المضاعفات
واهما احتمال الإجهاض من جراء سحب العينه 1%لثانية هي عن طريق سحب من
السائل الأمينوسي والموجود حول الجنين.وتأخذ هذه العينة في الأسبوع 14 -16
من الحمل واحتمال الإجهاض في هذه الحالة حوالي نصف في المائة 0.5%
فحوصات قبل الزواج
يمكن الكشف على أي شخص لمعرفة إذا ما كان حامل للمرض و بطريقة سهلة و ميسرة
و متوفرة في معظم المختبرات الطبية .و هو تحليل رخيص في قيمته المادة لكنه
غالي في قيمته العلمية. ولا يتوقع أن يكلف هذا التحليل أكثر من 15
دولار.و ينصح بإجراء هذا التحليل لكل من له أصول عرقية بمنطقة الخليج
العربي و جنوب السعودية و اليمن و المناطق الشمالية الغربية من السعودية. و
إن كنا نفضل أن يجرى هذا التحليل إضافة إلى تحاليل المتعلقة بالثلاسيميا
على جميع من يريد الزواج بغض النظر عن أصولة العرقية و ذلك لرخص قيمته و
توفره .
أمراض الدم الوراثية ,,
الثلاسيميــــــــــــــــا :
الثلاسيميا من أهم أمراض الدم الوراثية الانحلالية- التي تسبب تكسر كريات
الدم الحمراء- الشائعة على مستوى العالم بشكل عام وعلى مستوى منطقة البحر
الأبيض المتوسط بشكل خاص ، و الثلاسيميا نوعان الألفا و البيتا وحين يأتي
الحديث عن هذا المرض فإننا نقصد به (البيتا ثلاسيميا ).
و الثلاسيميا كلمه يونانية الأصل تعني فقر دم منطقة البحر الأبيض المتوسط
حيث أن هذا المرض عرف واشتهر في هذه المنطقة بشكل كبير و ، ويعرف أيضا باسم
أنيميا البحر المتوسط (Mediterranean anemia) وفي الولايات المتحدة
الأمريكية كان يعرف باسم أنيميا كوليز (Cooleys anemia).وفي منطقة البحر
الأبيض المتوسط كان لا بد أن يخضع كل مولود جديد بعد ولادته بشهور لهذا
الفحص لمعرفة ما إذا كان حاملا أو مصابا بالبيتا ثلاسيميا .أما في منطقة
الخليج العربي يحمل كل فرد من بين 20 فرد لهذا المرض أي نسبه حاملي صفة
المرض في الخليج 5 % وهي نسبه لا يمكن تجاهلها حيث أنه في كل مرة تحمل فيه
امرأة حاملة للمرض ومتزوجة من رجل أيضا حامل للمرض فإن احتمال إصابة
الجنين تصل إلى 25% وان اكثر من 60% من أطفال هذه الأسرة السليمين أيضاً
حاملين للمرض. وتكمن مشكلة المرض في عدم قدرة الجسم من تكوين كريات الدم
الحمراء - التي تنقل الغذاء والأكسجين إلى مختلف أنحاء الجسم – بشكل سليم
نتيجة لخلل في تكوين الهيموجلوبين( خضاب الدم) أدى إلا عدم اكتمال نضج
الكريه الحمراء و تكسرها وتحللها بعد فتره قصيرة من إنتاجها (يذكر أن عمر
كريه الدم الحمراء تكون 120 يوما) لذ فان المريض يحتاج إلى نقل دم بشكل
دوري كل 3-4 أسابيع حسب عمره و درجه نقص الهيموجلوبين .
ينتشر المرض في عدة مناطق من العالم ولكنها تكثر في المناطق التالية:
* حوض البحر الأبيض المتوسط ( اليونان، مالطا، قبرص، تركيا و إيطاليا).
* منطقة الخليج العربي.
* منطقة الشرق الأوسط وتشمل إيران العراق سوريا الأردن وفلسطين.
*شمال أفريقيا وتشمل مصر تونس الجزائر المغرب وبعض من الدول الأفريقية الأخرى.
* جنوب شرق أسيا وتشمل تايلاند، الفلبين، إندونيسيا، سنغافورة، كمبوديا، فيتنام وماليزيا.
* شبه القارة الهندية.
* دول أخرى كالصين، أرمينيا، جورجيا، أذربيجان.
المورث (الجين) المسؤول عن إنتاج مادة الألفا جلوبين: يسمى بمورث الألفا
جلوبين يوجد على الكروموسوم رقم 16 ،ويوجد منه أربع نسخ (أي أربع جينات او
مورثات)اثنتان منها موجودة على كروموسوم رقم 16 الأتي من الأم والاثنان
الآخران موجودان على الكروموسوم الأتي من الأب. واي خلل في هذه المورثات قد
يسبب مشاكل صحية حسب عدد المورثات المعطوبة او المصابة بالطفرة، واليك بعض
التفصيل
الألفا ثلاسيميا الساكتة Silent alpha Thalassemia):
فإذا أصابت الطفرة مورث واحد فقط فأن الشخص لا يصاب بأي مشكلة صحية و تسمى
هذه الحالة بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia).وفي
العادة لا يعرف الشخص انه حامل لمورث معطوب وقد يصعب التعرف على هذه الحالة
حتى عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا
يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر
الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر إلا بالكشف
على المورثات مباشرة،وهذه غير متوفرة في الكثير من المراكز الطبية.
الحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait)
فلو أصاب العطب مورثين من هذه الأربعة فقط سمي الشخص حامل لصفة الألفا
ثلاسيميا وهو لا يصاب بأية مشكل صحية ولكن قد يولد له أطفال مصابون بمشاكل
صحية في الهيموجلوبين إذا كانت زوجته حاملة لنفس الصفة.
مرض بالهيموجلوبين اتش :
لو أصاب العطب(الطفرة)ثلاث مورثات من الأربعة يصاب الشخص بمرض يسمى
بالهيموجلوبين اتش (Hemoglobin H disease) .وعند ولادة الطفل المصاب بعطب
في ثلاثة مورثات من نوع ألفا يكون لدية قلة في كمية الهيموجلوبين في كريات
دمهم الحمراء مع وجود هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج
عن اتحاد أربع قطع من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة
التي تقل فيها كمية الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من
النوع المتوسط فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100
ملليتر ويكون لدية تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال
لنقل الدم، أما البالغين فقد يحتاجون لنقل كميات من الدم.
استسقاء شديد للجنين (Hydrops Fetalis):
لو أصاب العطب(الطفرة)جميع المورثات الأربعة يصاب الجنين وهو في بطن أمه
باستسقاء شديد في جميع الجسم نتيجة لوجود فقر دم حاد مع فشل في القلب
كنتيجة ثانوية لنقص الهيموجلوبين في الدم.
المورث (الجين) المسؤول عن إنتاج مادة البيتا جلوبين: يسمى بمورث البيتا جلوبين يوجد على الكروموسوم 11.
حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى)
تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب.
وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا
يعاني حامل المرض من أي مشكلة صحية إلا الهم من فقر دم طفيف ولا يحتاج إلى
نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك
نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من
السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد
صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك
إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا
شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا
حصيات في المرارة أو تقرح في ساق القدم.
حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى)
تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب.
وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا
يعاني حامل المرض من أي مشكلة صحية إلا الهم من فقر دم طفيف ولا يحتاج إلى
نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك
نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من
السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد
صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك
إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا
شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا
حصيات في المرارة أو تقرح في ساق القدم.
مصاب بالثلاسيميا المتوسط
تحدث الثلاسيميا المتوسطة عند وجود عطب(طفرة) في كلا مورثي البيتا جلوبين .
فينتج عن ذلك نقص متوسط الشدة في كمية البيتا جلوبين المنتجة في الجسم
وتؤدي في هذه الحالة إلى نقص متوسط الشدة لمستوى الهيموجلوبين في الدم
فيتراوح في العادة بين 7-10 غرام لكل 100 ملليمتر ،وفي العادة لا يحتاج
المريض لنقل دوري للدم ، لكنه مع التقدم في العمر و في خلال الحمل في حالة
المرأة قد يحتاج إلى نقل دم وأشراف طبي بشكل منتظم. و تشكل الثلاسيميا
المتوسطة من 2-10% من مرضى البيتا ثلاسيميا ،وتظهر أعراضها خلال ألسنه
الثانية إلى الرابعة من عمر المريض ، و مخبرياً تظهر علامات فقر الدم
خلال السنة الأولى من عمر المريض من خلال فحص الدم الروتيني.وقد يتعرض
المريض لبعض المشاكل الصحية كضعف البنية وتضخم الكبد والطحال و الإصابة
باليرقان(اصفرار الجلد والعينان).
مصاب بالثلاسيميا (الثلاسيميا الكبرى)
تحدث الثلاسيميا الكبرى أو الشديدة عند وجود عطب(طفرة) في كلا مورثي
البيتا جلوبين كما هو الحال في الثلاسيميا المتوسطة ولكن نوع العطب
(الطفرة)في مورث البيتا هذه المرة اشد فينتج عن ذلك نقص شديد نسبة البيتا
جلوبين فينقص بذلك الهيموجلوبين إلا نسب اقل من 7 غرام لكل 100 ملليليتر
نتيجة لتكسر كريات الدم الحمراء الغير طبيعية قبل انتهاء عمرها الافتراضي
(120 يوم). و يحتاج المريض لنقل دوري للدم كل 3-4 أسابيع للمحافظة على نسبه
عالية من الهيموجلوبين لكي ينموا الجسم بشكل صحي. ويمكن اكتشاف المرض خلال
الأشهر الستة الأولى من عمر الطفل
مريض بالثلاسيميا (الثلاسيميا الكبرى) + الأنيميا المنجلية
في هذه الحالة يصاب المريض بمرضين في أن واحد.فيكون الشخص أما لدية مرض
الأنيميا المنجلية ومرض الثلاسيميا معا أو يكون حاملا للأنيميا المنجلية مع
مرض الثلاسيميا.وقد تتراوح شدة المرض وانقص الهيموجلوبين على حسب شدة
الإصابة في مورث البيتا جلوبين.
الاعـــــــــراض :
تختلف أعراض المرض على حسب نوع الثلاسيميا المصاب بها الشخص.وقد لا يشعر
المصاب بالمرض بأي مشكلة كما هو الحال فالشخص الحامل للألفا ثلاسيميا
وتكتشف بالمصادفة عند إجراء تحليل دم لكريات الدم الحمراء والهيموجلوبين.و
في المقابل قد يكون شديد كما هو الحال في البيتا ثلاسيميا الكبر في تكون
الأعراض واضحة و تتفاقم المشكلة إذا لم ينقل للمريض دم بشكل دوري.كما أن
المرض قد يكون شديد لدرجة أن الجنين يموت في بطن أمة أو يولد مع استسقاء في
جميع الجسم نتيجة لنقص الهيموجلوبين في الدم وهبوط شديد في القلب،وهذا
يحدث في الألفا ثلاسيميا التي يكون فيها جميع مورثات(جينات)الألفا
هيموجلوبين معطوبة (بها طفرة مرضية).
ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال
وفيما يلي الأعراض التي تحدث للمصابين بمرض البيتا ثلاسيميا الكبرى إذا لم يعالج المريض:
.شحوب واصفرار البشرة والشفتين
الخمول و الشعور بالتعب والإرهاق لأقل جهد.
فقدان الشهية.
.نقص حاد في الهيموجلوبين (اقل من 9 مج\مل دم)
تضخم الكبد والطحال نتيجة عجز نخاع العظام عن إنتاج كريات الدم الحمراء
فيبدأ الكبد والطحال وأعضاء أخرى بمحاولة تصنيع الدم بنفسها مما يؤدي إلى
تضخمها.
الانعكاسات النفسية السيئة لدى المريض نتيجة شعوره بالمرض الدائم والنقص الصحي مقارنة بأقرانه.
التأخر في النمو الجسماني كالطول والوزن .
تغيرات في عظام الجسم ومنها الجمجمة ، بروز الجبهة وعظام الوجنتين ، انخفاض عظام الأنف وبروز عظام الفك العلوي.
زيادة الالتهابات بشكل عام.
سرعة ضربات القلب لمحاولة تعويض نقص الهيموجلوبين وذلك بزيادة سرعة ضخ القلب للدم، كذلك قد تحدث مضاعفات قلبيه مع مرور الوقت.
حدوث مضاعفات نتيجة تراكم الحديد في أعضاء مختلفة من الجسم أن لم ينتظم
المريض باستعمال الحقنه اليومية للدسفيرال والتي تؤدي إلى التخلص من الحديد
عن طريق البول وتتمثل هذه المضاعفات في تشحم الكبد ، اسوداد لون الجلد،
خلل هرموني (مرض السكري) ، انخفاض في هرمون الغدة الدرقية وهرمون الجار
درقية وهرمونات الغدة النخامية
مشاكل في القلب كتضخم عضلة القلب مع هبوط في القلب وعدم أنتظام في دقات القلب .
احتمال أصابه المريض بالأمراض المعدية نتيجة لنقل الدم بشكل مستمر و لكن
قلة تلك المخاطر بعد قيام جميع مراكز التبرع بالدم بفحص إذا ما كان دم
المتبرع بدم مصاب بالإيدز او فيروسات التهاب الكبد الوبائي.
الاسبـــــــاب :
الثلاسيميا مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض
أطفالهم.ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور
المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.ولكي تفهم هذا
الجانب العلمي أنصحك بالرجوع إلى صفحة أساسيات الوراثة لمعرفة ما هي
الخلية والنواة و الكروموسوم والمورث .وسوف أقوم بشرح مختصر عن هذا الكلمات
في سياق كلامي عن الهيموجلوبين والمورثات في هذه الصفحة.
تصنيع الهيموجلوبين:
يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع
العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها
البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم
(Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى
بالجلوبين(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع
بيتا وفي هذه الحالة يسمى هذا الهيموجلوبين بهيموجلوبين ( أ ) أو (A ).
هذا الترابط العجيب بين الحديد و الهيم و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم.
كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي
عبارة عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل
الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات(الصبيغات)
،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين
يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات
موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض
الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة)
في المورثات التي تصنع مادة الجلوبين.
المورثات و الجلوبين:
هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى
تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان
من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على
الكروموسومات،كما هو الحال في مادة الهيم.
ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم
11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11
الذي ورثة الإنسان من أمة..بينما يصنع مادة الألفا جلوبين من أربع
مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين
على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من
الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان
بمرض ألفا ثلاسيميا، و إذا أصاب الخلل مورث من مورثات البيتا يصاب الإنسان
بمرض البيتا ثلاسيميا.هذه بشكل مبسط العلاقة بين المورثات و
الثلاسيميا،ولكن الموضوع اعقد من ما ذكرنا ،ولكي تعرف المزيد تابع القراءة!
البيتا ثلاسيميا (Beta Thalassemia ) :
خلاصة ما ذكرنا أن هناك أربع مورثات لتصنيع مادة الألفا جلوبين، و مورثين
لتصنيع مادة البيتا جلوبين.واليك الآن بعض التفصيل في هذا الأمر و لنبدأ
بالبيتا ثلاسيميا.لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات
البيتا جلوبين فأن الخلية تستمر في إنتاج البيتا جلوبين وذلك نظرا لوجود
مورث أخر سليم .ولكن هناك نقص في الكمية الإجمالية المنتجة من هذه المادة
ولكن هذا النقص لا يسبب مرضا ولكن يسمى الشخص الذي يحمل مورث واحدا معطوبا
من مورثات البيتا بالحامل للمرض،أي انه حامل لمرض البيتا ثلاسيميا ويمكن
أن يتعرف على هذه الحالة بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)وهي
متوفرة في الكثير من المستشفيات.أما لو حدثت الطفرة في كلا النسختان فان
مجوع الكمية المصنعة من البيتا جلوبين تقل بشكل كبير فيصاب الشخص بمرض
البيتا ثلاسيميا وتكون شدة المرض على حسب شدة الطفرة فبعض الطفرات تسمح
بإنتاج كمية معقولة من مادة البيتا جلوبين(ويسمى المرض في هذه الحالة
بالبيتا ثلاسيميا الصغرى)وهناك طفرات شديدة تؤدي إلى نقص كبير وقد يكون
كامل في مادة البيتا جلوبين (ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا).
نظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض
على حسب عدد المورثات المعطوبة .فإذا أصيب مورث واحد فأن الشخص لا يصاب
بأي مشكلة صحية(يسمى بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia)
وقد يصعب التعرف على هذه الحالة عند إجراء تحليل دم عادي(أي عدد الكريات
الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة
الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه
يصعب اكتشاف هذا الأمر الا بالكشف على المورثات مباشرة،وهذه غير متوفرة في
الكثير ن المراكز الطبية.بينما فحص حركة الهيموجلوبين الإلكترونية (
Hemoglobin Electrophoresis) متوفر في الكثير من المستشفيات.
الالفا ثلاسيميا ( Alpha thalassemia):
بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم
16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم
16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة)
في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا.
ونظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض
على حسب عدد المورثات المعطوبة .فإذا أصابت الطفرة مورث واحد فقط فأن
الشخص لا يصاب بأي مشكلة صحية و تسمى هذه الحالة بالألفا ثلاسيميا الساكتة
Silent alpha Thalassemia).وفي العادة لا يعرف الشخص انه حامل لمورث معطوب
وقد يصعب التعرف على هذه الحالة حتى عند إجراء تحليل دم عادي(أي عدد
الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة
الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه
يصعب اكتشاف هذا الأمر إلا بالكشف على المورثات مباشرة،وهذه غير متوفرة في
الكثير من المراكز الطبية.أما إذا أصابت الطفرة مورثين اثنين من الأربعة
فان هذه الحالة تسمى بالحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia
Trait) وهذا الشخص أيضا لا يصب بمشاكل في الصحة وليس علية خطورة .ولكن
يمكن الاشتباه في أن الشخص لدية هذا النوع من الثلاسيميا عند إجراء تحليل
عادي لكريات الدم الحمراء ،ففي هذه الحالة تكون كريات الدم الحمراء اصغر من
الحجم المعتاد وكمية الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات
التي على كريات الدم الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال
المصابون بفقر الدم الناتج عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس
كمية الحديد في الدم فإذا كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن
الألفا ثلاسيميا .أما إذا كان هناك نقص في الحديد فيعالج الأمر بإعطاء
الحديد ثم يعاد التحليل .و إذا زال نقص الحديد واستمر صغر كريات الدم
الحمراء فان هذا يعني أن الشخص كان عنده نقص في الحديد مصحوب بألفا
ثلاسيميا.يكثر الاشتباه بين فقر الدم الناتج عن نقص الحديد و ألفا ثلاسيميا
لذلك على العائلة تنبيه الطبيب على أن بعض أفراد العائلة لديهم ألفا
ثلاسيميا لكي لا يعطى الطفل أو البالغ(كامرأة الحامل) جرعات من الحديد من
دون أن يكون هناك ضرورة لهذا الأمر لأن ارتفاع كمية الحديد في الجسم
مضر.أما إذا أصاب العطب(الطفرة)ثلاث مورثات من الأربعة فان الشخص يصاب بمرض
يسمى بمرض الهيموجلوبين اتش (Hemoglobin H disease).وعند ولادة الطفل
المصاب يكون هناك نقص في كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود
هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع
من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية
الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من النوع المتوسط
فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية
تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال لنقل الدم، أما
البالغين فقد يحتاجون لنقل كميات من الدم.أما إذا أصاب العطب(الطفرة)جميع
المورثات الأربعة يصاب الجنين باستسقاء شديد في جميع الجسم نتيجة لوجود فقر
دم حاد مع فشل في القلب كنتيجة ثانوية لنقص الهيموجلوبين في الدم.ويموت
الجنين إذا لم ينقل له جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل
دم بشكل دوري نتيجة لوجود فقر دم شديد إلى متوسط الشدة.
التشخيــــــــص :
الهيموجلوبين وفقر الدم الناتج عن تكسر الدم:
الهيموجلوبين عبارة عن بروتين موجود في كريات الدم الحمراء وظيفته نقل
الأكسجين من الرئة إلى كافة خلايا الجسم ،وهو يتكون من الهيم (مادة الحديد +
صبغه) و الجلوبين (البروتين). وهناك أنواع مختلفة من بروتينات الجلوبين
المهمة منها الألفا ،البيتا ، الدلتا و الجاما جلوبين .وتحدث المشكلة
عندما يحدث نقص في مكونات بروتين الجلوبين بسبب خلل جيني وكل زوج من
الجينات الآتية من الأم و الأب يعمل على تنظيم و ناتج بروتينات الجلوبين
في الجسم فمثلا إذا كان الجين المسؤول عن بروتينات البيتا جلوبين به خلل
فلن يكون ناتج التكوين كاملا وصحيحا مما يؤدي إلى خلل في تكوين سلاسل
البيتا وبالتالي يؤدي إلى مرض البيتا ثلاسيميا. كما انه اذا كان نفس الخلل
موجود في بروتين الألفا جلوبين فانه ينتج مرض الألفا ثلاسيميا. وكلا
الألفا و البيتا ثلاسيميا يعتبران من الأمراض الشائعة جدا في منطقة الشرق
الأوسط وبأنواع نادرة أيضاويتغير نوع الهيموجلوبين في الجسم خلال الحمل إلى
عدة شهور بعد الولادة نتيجة لتغير نوع الجلوبين المنتج:
فعند الولادة يكون اغلب الهيموجلوبين(بنسبة تزيد عن 80%) من ما يعرف باسم
الهيموجلوبين الجنيني ( Hb F) ويستمر تواجده إلى أن تتم عملية إنتاج
الهيموجلوبين البالغين( Hb A) بشكل منتظم خلال الستة شهور الأولى من
العمر.,كما يوجد نسبة قليلة (1.5-2.5%) من هيموجلوبين يسمى بالهيموجلوبين
البالغين نوع 2 (( Hb A2) ).
المعدل الطبيعي للهيموجلوبين عند البالغين الذكور = 13-16 جرام/ 100 ملليليتر وعند البالغات الإناث = 12-15 جرام/ 100ملليليتر .
حامل البيتا ثلاسيميا:
يصعب التاكد من ان الشخص حامل لثلاسيميا و لكن في العادة يكون مستوى
الهيموجلوبين طبيعي ولكن كريات الدم الحمراء اصغر من المعدل الطبيعي( MCV
اقل من 75).و عند اجراء فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin
Electrophoresis) يكون نسبة الهيموجلوبين A2 و هيموجلوبين F اعلى من
الطبيعي .
مصاب بالبيتا ثلاسيميا:
يكون مستوى هيموجلوبين في الدم اقل من 10 مليغرام لكل 100 مليلتر . و تكون
كريات الدم الحمراء اصغر من المعدل الطبيعي( MCV اقل من 75).و عند اجراء
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) يكون
نسبة الهيموجلوبين A2 و هيموجلوبين F اعلى من الطبيعي .
الألفا ثلاسيميا الساكتة Silent alpha Thalassemia):في العادة يصعب
التعرف على هذه الحالة فتحليل الدم طبيعي(أي عدد الكريات الحمراء
والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين
الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف
هذا الأمر إلا بالكشف على المورثات مباشرة (اي فحص DNA) ،وهذه غير متوفرة
في الكثير من المراكز الطبية.
حامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait) وهذا الشخص لا يصاب
بمشاكل في الصحة وليس علية خطورة .و لكن يمكن الاشتباه في أن الشخص لدية
هذا النوع من الثلاسيميا عند إجراء تحليل عادي لكريات الدم الحمراء ،ففي
هذه الحالة تكون كريات الدم الحمراء اصغر من الحجم المعتاد وكمية
الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات التي على كريات الدم
الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال المصابون بفقر الدم الناتج
عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس كمية الحديد في الدم فإذا
كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن الألفا ثلاسيميا .أما إذا
كان هناك نقص في الحديد فيعالج الأمر بإعطاء الحديد ثم يعاد التحليل .و
إذا زال نقص الحديد واستمر صغر كريات الدم الحمراء فان هذا يعني أن الشخص
كان عنده نقص في الحديد مصحوب بألفا ثلاسيميا.يكثر الاشتباه بين فقر الدم
الناتج عن نقص الحديد و ألفا ثلاسيميا لذلك على العائلة تنبيه الطبيب على
أن بعض أفراد العائلة لديهم ألفا ثلاسيميا لكي لا يعطى الطفل أو
البالغ(كامرأة الحامل) جرعات من الحديد من دون أن يكون هناك ضرورة لهذا
الأمر لأن ارتفاع كمية الحديد في الجسم مضر.قد يساعد تحليل حركة
الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) يكون نسبة
الهيموجلوبين A2 و هيموجلوبين F اعلى من الطبيعي .
مرض الهيموجلوبين اتش (Hemoglobin H disease) والذي يحدث نتيجة لوجود عطب
في ثلاث نسخ من الالفا جلوبين.عند ولادة الطفل المصاب يكون هناك نقص في
كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود هيموجلوبين غير طبيعي
يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع من جلوبين جاما .وبعد
مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية الجاما جلوبين في جسم
الإنسان) يصاب الطفل بفقر دم من النوع المتوسط فيتراوح مستوى الهيموجلوبين
حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان .
عطب أربع نسخ من مورثات الالفا جلوبين(استسقاء شديد للجنين): و يحدث
الاستسقاء نتيجة لوجود فقر دم حاد مع فشل في القلب كنتيجة ثانوية .مستوى
الهيموجلوبين منخفض.و في العادة يموت الجنين او يولد ميت إذا لم ينقل له
جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل دم بشكل دوري نتيجة لوجود
فقر دم شديد إلى متوسط الشدة. يمكن الكشف عن المرض باجراء حركة
الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) و يلاحظ عدم و جود
اي نسبة من هيموجلوبين A و لا A2 و لا F
العــــــــــــــلاج :
وسائل علاج البيتا ثلاسيميا:
نقل دم
ينقل الدم بشكل دوري للمريض (6-8 ملليليتر دم"كريات دم حمراء" لكل كجم من
وزن المريض كل 3-4 أسابيع)، ويجب ان يكون عمر الدم المنقول لا يتجاوز ال6
أيام. والنقل الدوري يحافظ على مستوى عالي من الهيموجلوبين لكي يصل
الأكسجين الى اجزاء الجسم بشكل افضل فيحافظ على النمو الطبيعي للطفل، ومنع
التغيرات التي تحدث في عظام الجسم،حماية القلب من مضاعفات فقر الدم، منع
تضخم الكبد والطحال.
ويحتاج المريض إلى كريات الدم الحمراء فقط لذا يجب ان يرشح الدم المنقول من الكريات البيضاء و الصفائح الدموية.
العلاج بالديسفسرال
يترسب الحديد في جسم المريض تدريجيا مما يؤدى إلى تعطيل وظائف الخلايا
وبالتالي إلى موتها وفقدان وظائفها، كما يؤدى إلى الكثير من اختلال الوظائف
الهرمونية و القلبية و الكبدية و الجلدية.
لذا يجب التخلص من الحديد الزائد (اكثر من 1000 مج\مل)
الديسفرال عبارة عن مادة ترتبط مع الحديد في الجسم وتخرج مرتبطة بالحديد
لخارج الجسم عن طريق البول، ومن هنا تأتى أهميه الديسفرال الذي يحقن عن
طريق الجلد لفتره8-10 ساعات يوميا او عن طريق الوريد،والعضل
إزالة الطحال
للطحال دور هام في التخلص من كريات الدم الميتة نتيجة مشاركة الطحال في
تعويض الجسم عن عجز نخاع العظم واستمرار تراكم الحديد بصوره كبيره يبدأ
بعدها الطحال بتدمير كريات الدم الحمراء والبيضاء والصفائح الدموية ،تتفاقم
حدة فقر الدم وتتناقص مناعة ومقاومة المريض ويصبح عرضة للنزف، ويتم
استئصال الطحال عندما تظهر المؤشرات التالية:
عندما يحتاج المريض إلى نقل كميات من الدم تزيد بمقدار واحد ونصف عن الكميه الكافية.
نقص حاد في كريات الدم البيضاء أو الصفائح الدموية نتيجة تدميرها في الطحال
التضخم الكبير والمتزايد للطحال.
زرع نخاع العظم
هذه العملية تعتمد على وجود متبرع يفضل أن يكون من أشقاء او شقيقات المريض
حيث يجب أجراء الفحوصات التي تؤكد من وجود التطابق النسيجي والخلوي (100%)
بين المتبرع والمريض،وتزداد فرص نجاح العملية للذين لا يعانون من ترسب
الحديد أو تضخم الكبد والطحال.
إن رفض الانسجه عادة ما يكون اقل نسبة في الأطفال عنه في الكبار فذلك لا
يحدث مباشرة بعد العملية إنما قد يحتاج إلى 3-5 سنوات يبقى فيها خطر
الرفض،فالمريض بحاجة إلى متابعة دقيقه للغاية.
ويتم فبل زرع نخاع العظام التخلص من نخاع المريض بتناوله الأدوية تدمر
خلايا النخاع وعلى مدى 6 أيام يشعر فيها بالوهن والضعف وتحت التعقيم التام
يتم شفط النخاع بواسطة حقنه خاصة وتحت التخدير العام وتستغرق هذه العملية
حوالي نصف الساعة وتستكمل العملية بإعادة حقن نخاع العظام السليم إلى
المريض فيما يشبه عملية نقل الدم فيجد النخاع الجديد تجويف نخاع المصاب.
والمتبرع عادة لا يعاني من أي مخاطر وجسمه قادر على تعويض الكميه التي تم
شفطها من نخاع العظام ولا يحتاج للبقاء في المستشفى اكثر من يوم واحد فقط.
الجديد في علاج البيتا ثلاسيميا:
1- بدأت بعض المراكز في إجراء عمليات نقل وزراعة نخاع العظم للجنين المصاب
داخل رحم أمه بدل إجهاضه وتتميز هذه العملية بندرة رفض جسم المريض للنخاع
المزروع.
2- يتوفر الان نوع مشابه لدواء الدسيفيرال الطارد للحديد من الجسم و يأخذ
عن طريق الفم بدل من الحقن التحت جلدية. و الابحاث المتتالية اثبتت فعالية
هذا الدواء مقارنة بالديسفيرال االذي يؤخذ عن طريق الحقن و يسمى هذا الدواء
الديفريبرو ن (deferiprone)و يسوق تحت اسم تجاري يدعى Ferriprox™.
طرق الوقاية:
فحوصات قبل الولادة
إذا كان كلا الوالدان يحملان صفة البيتا ثلاسيميا الصغرى فمن الممكن أن يتم
فحص الجنين للتأكد من سلامته من مرض الثلاسيميا خلال الأسبوع 10-11 من
الحمل. ويتم الفحص بأخذ عينه من المشيمة ( Chorionic Villus )وتحليلاها
بواسطة ال DNA لمعرفة إذا ما كان الجنين مصاب بالثلاسيميا أم لا.ولكنا يجب
أن يحضر لهذا الأمر فيجب أولا معرفة نوع الطفرة الموجودة في العائلة لكي
يستطيع الأطباء الكشف عنها خلال الحمل،وتظهر نتيجة التحليل خلال 3-4
أسابيع.وكما هو الحل في جميع الفحوصات الطبية فقد يكون هناك بعض المضاعفات
واهما احتمال الإجهاض من جراء سحب العينه 1%لثانية هي عن طريق سحب من
السائل الأمينوسي والموجود حول الجنين.وتأخذ هذه العينة في الأسبوع 14 -16
من الحمل واحتمال الإجهاض في هذه الحالة حوالي نصف في المائة 0.5%
فحوصات قبل الزواج
يمكن الكشف على أي شخص لمعرفة إذا ما كان حامل للمرض و بطريقة سهلة و ميسرة
و متوفرة في معظم المختبرات الطبية .و هو تحليل رخيص في قيمته المادة لكنه
غالي في قيمته العلمية. ولا يتوقع أن يكلف هذا التحليل أكثر من 15
دولار.و ينصح بإجراء هذا التحليل لكل من له أصول عرقية بمنطقة الخليج
العربي و جنوب السعودية و اليمن و المناطق الشمالية الغربية من السعودية. و
إن كنا نفضل أن يجرى هذا التحليل إضافة إلى تحاليل المتعلقة بالثلاسيميا
على جميع من يريد الزواج بغض النظر عن أصولة العرقية و ذلك لرخص قيمته و
توفره .

DREAM MAN- مدير موقع المعهد الطبي

- تاريخ التسجيل : 25/01/2010
عدد المساهمات : 533
المزاج : رايق
الجنس :
رد: امراض الدم الوراثية بالتفصيل و بالعربي
من طرف DREAM MAN الخميس يناير 27, 2011 11:48 pm
الانيميـــــــــا المنجليـــــــــة :
الانيميا المنجلية هي احد أنواع فقر الدم.وهي تصيب كريات الدم الحمراء.و هي
من اشهر أمراض الدم الوراثية الانحلالية- التي تسبب تكسر كريات الدم
الحمراء-و هي اكثرها شيوعاً على مستوى العالم بشكل عام و في افريقيا و
الشرق الاوسط بشكل خاص . و هناك عدة اسماء لهذا المرض ،فاضافة الى الانيميا
المنجلية فانه يطلق عليها ايضا فقر الدم المنجلي او مرض المنجلية.وايا كان
الاسم فالمرض واحد و ليس هناك فروق بينها.
و كلمة المنجلية مأخوذه من المنجل(الذي يحصد به النبات و في بعض المناطق
يطلق علية المحش)و ذلك لان كريات الدم الحمراءتحت المجهر تأخذ شكل مقوس
كالمنجل او الهلال .وكلمة انيميا تعني فقر دم .و ينتشر المرض في عدة دول
افريقية و اسيوية.وهو تقريبا موجود في جميع الدول العربية .أما في بعض
مناطق من الخليج العربي (كمنطقة الاحساء و القطيف و جازان في
السعودية)فيحمل كل 4 افراد من بين 20 فرد هذا المرض، أي نسبه حاملي صفة
المرضحوالي 20 % ،و هي نسبه لا يمكن تجاهلها حيث أنه في كل مرة تحمل فيه
امرأة حاملة للمرض ومتزوجة من رجل أيضا حامل للمرض فإن احتمال إصابة الجنين
تصل إلى 25% وان اكثر من 60% من أطفال هذه الأسرة السليمين أيضاً حاملين
للمرض. وتكمن مشكلة المرض في انتاج نخاع العظم لكريات دم حمراء - التي تنقل
الغذاء والأكسجين إلى مختلف أنحاء الجسم – غير طبيعية نتيجة لخلل في تكوين
الهيموجلوبين( خضاب الدم) .وهذه الخلايا غير الطبيعية تأخذ شكل المنجل و
هي قابلة الى التكسر وتتحلل بعد فتره قصيرة من إنتاجها و قد تعيق مرور
الدم خلال الشعيرات الدموية، و قد تسد عروق الدم فتسبب الام مبرحه في
اجزاء مختلفة من الجسم خاصة في العظام خاصة عظام الاطرف و الظهر.و قد تسد
كريات الدم الحمراء المنجلية اي عرق من العروق الدموية في الرئتين او في
البطن او حتى في المخ و قد تسبب مضاعفات خطيرة اضافة الى الألام المببرحه
التي يعاني منها الشخص المصاب. .
ينتشر المرض في عدة مناطق من العالم ولكنها تكثر في المناطق التالية:
* افريقيا بشكل عام.
* منطقة الخليج العربي و في اليمن و جنوب شرق السعودية.
* منطقة الشرق الأوسط وتشمل إيران العراق سوريا الأردن وفلسطين.
* شبه القارة الهندية.
* جنوب شرق أسيا .
* المنطقة الكاريبية في امريكا الوسطى
الحامل للانيميا المنجلية (Sickle cell trait) :
يكون الشخص حامل للانيميا المنجلية اذا وجود مورث سليم لسلسة البروتين بيتا
و أخر معطوب،ولا يعاني حامل المرض في العادة من أي مشكلة صحية. و يمكن
التعرف على الشخص الحامل عن طريق فحص بسيط للدم.
المصاب بالانيميا المنجلية:
هو شخص لديه عطب في كلا الجينين المصنعين لسلسة البروتين بيتا ( Beta
Globin chain).و عند الولادة يكون الطفل سليم و لكن عند بلوغ الشهر السادس
او اكثر من العمر يبدأ ظهور علامات المرض و التي في العادة تكون مصحوبة
بانخفاض بنسبة الهيموجلوبين(فقر دم)مع شحوب في البشرة و انتفاخ في اليدين و
القدمين و بكاء ناتج عن الام في العظام.و قد تكون حالة الطفل حرجة اذا كان
مصحوب بالتهاب بكتيري في الدم.و في العادة يتراوح مستوى الهيموجلوبين
حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان .
ويحتاج الطفال لنقل الدم اذا انخفض الهيموجلوبين اقل من 7 غرامات او كانت .
مصاب الأنيميا المنجلية و بالثلاسيميا
في هذه الحالة يصاب المريض بمرضين في أن واحد.فيكون الشخص أما لدية مرض
الأنيميا المنجلية ومرض الثلاسيميا معا أو يكون حاملا للأنيميا المنجلية مع
مرض الثلاسيميا.وقد تتراوح شدة المرض وانقص الهيموجلوبين على حسب شدة
الإصابة في مورث البيتا او الالفا جلوبين.و فيالعادة تكون الاصابة في
الشخاص المصابين بالانيميا المنجلية و البيتا ثلاسيميا اقل شدة ممن يصاب
بالانيميا المنجلية لوحدها و ذلك حدوث الثلاسيميا يقلل من الكمية المنتجة
من الهيموجلوبين المنجلي و الذي يسبب مشاكل كثيرة و يسد الشعيرات الدموية.
المورث (الجين) المسؤول عن إنتاج مادة البيتا جلوبين: يسمى بمورث البيتا
جلوبين يوجد على الكروموسوم 11. و كل انسان لديه نسختان من هذا المورث
،نسخة تاتي من الاب و الاخرى من الام
الأعــــــراض :
لا تظهر اي اعراض على الشخص الحامل للانيميا المنجلية و قد تكتشف
بالمصادفة عند إجراء اختبار التمنجل والذي يجرى في العادة قبل اجراء عملية
جراحية حيث يجرى بشكل روتيني قبل العملية في كثير من البلدان.
اما بنسبة للمصاب الانيميا المنجلية فالاعراض تختلف على حسب سن المريض:
قبل الشهر السادس من العمر:
لا تظهراي اعراض خلال الاشهر الاولى من العمر حتى في الشخص المصاب
بالانيميا المنجلية.وهذا لان نخاع العظم(المصنع الذي ينتج كريات الدم
الحمراء)لا يبدأ في صناعة الهيموجلوبين الذي يحتاج الى البيتا جلوبين الى
بعد مضي عدة اشهر من الولادة.و كما شرحنا في قسم الاسباب فان البيتا جلوبين
تدخل في تكوين الهيموجلوبين S .
بعد الشهر السادس و خلال السنتين الاوليين من العمر:
من اهم الاشكال التي يظهر فيها المرض في هذا السن ما يعرف مرض اليدين
والقدمين.حيث تنتفخ اليدين و القدمين مصحوب بالم و بكاء .و قد يتركز التورم
في الاصابع . كما يكون بشرة الطفل شاحبة نتيجة لوجود فقر الدم.
في بعض الاحيان تكون هذه الحالة مصحوبة بالتهاب فيروسي او بكتيري في
الدم.وقد يكون هذا الالتهاب شديد و قد يستدعي ان يدخل الطفل العناية
المركزة.
بعدها تبدأ حدوث النوبات المتكررة من الالم و الناتج لانسداد العروق
الدموية المغذية للعظام خاصة عظام الاطراف و العمود الظهري.تختلف شدة هذه
النوبات و عدد مرات تكرارها من شخص الى اخر و يبدوا ان هناك نوعان من
الانيميا المنجلية .نوع يكون شديد و يتركز هذا في المنطقة الجنوبية الغربية
من السعودية (جازان و ماحولها من المناطق)و نوع اقل شدة في الحالات التي
يكون مصدرها من المنطقة الشرقية من السعودية.اتفاوت الشدة غير معروف السبب
بشكل كامل.و لكن هناك تكهنات بان المرض يكون قل خطورة كلما كانت نسبة
الهيموجلوبين F عليه.و قد يكون مستوى الهيموجلوبين F في الحالات التي في
المنطقة الشرقية من السعودية اعلى من الحالات التي يكون مرجعها المنطقة
الجنوبية من السعودية.هناك رأي اخر و ذلك يرجع الى الاصول الوراثية للاشخاص
المصابين بهذا المرض.فالمرض الذي في المنطقة الجنوبية يسمى بالبينين (
Benin) ويله اصول افريقية.بينما الذي في المنطقة الشرقية فيعرف بالسعودي
الهندي و ذلك لتشابهه للحالات التي في الهند.
خلا ل سنتين الى العاشرة سنوات من العمر:
في معظم الوقت تكون البشرة شاحبة نتيجة لوجود فقر الدم حيث يتراوح مستوى الهيموجلوبين بين 7-10 مليغرام.
اصفرار البشرة و العينين نتيجة لتكسر الدم و ارتفاع المادة الصفراء في الدم(يرقان)
اقد تكون البنية نحيفة ،مع تعب و ارهاق عند القيام باعمال فيها جهد عالي.
نوبات الالم.و سوف نفصل في هذه النوبات في اخر هذه الصفحة .
زيادة القابلية للاصابة بالالتهابات خاصة الالتهابات البكتيرية (خاص
Hemophilus Infeluanza و Pnueomococus ) وي مكن الوقاية منها باخذ القاح
الواقي منها(تطعيم).
تكثر الالتهابات افي الجهاز النفسي، و في بعض الاحيان يحدث التهاب في العظم
. و قد يلتبس او يصعب على الاطباء في هذه الحالة التمييز بين نوبة الالم
المتكرة الحدوث وبين التهاب العظم .وهذا مشهور بين الاطباء.
بعد سن العاشرة من العمر:
ان اهم ما يميز هذا العمر هو زيادة احتمال اصابة الرئتين بتجلطات نتيجة
لانسددا العروق الدموية بالخلايا المنجلية.و تظهر هذه الحالات على شكل الم
في الصدر مع كحة جافة و نقص في الاكسجين. و قد تلتبس او يصعب على الاطباء
التفرقه بينها و بين الالتهاب الرئوي.
شحوب مصحوب ببشرة داكنة مع بروز في عظام الجبهة والوجنتين والفك العلوي.مع
ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال
مشاكل صحية قد تحدث في اي عمر:
نوبات الم
التهابات متكررة
تقرح في الساقين يصعب شفائها
حصوات في المراراة.
انتصاب مؤلم عند الذكور (Priapism) .
فقر الدم الناتج عن نقص حمض الفوليك. و قد يظهر هذا على شكل انخفاض في
نسبة الهيموجلوبين مع كبر في حجم الخلايا الحمراء . وقد يتطور و تبداء
الخلايا البيضاء في الانخفاض لو كان النقص شديد. و الاطباء يعطون جميع
المصابين بالانيميا المنجلية حمض الفوليك بشكل يومي و ذلك لزيادة الحاجة
لهذا الفيتامين المهم في تكوين و صنع خلايا الدم خاصة الخلايا الحمراء.
حدوث شلل نصفي نتيجة لاتسداد احد العروق في المخ.يقوم الكثير من الاطباء
الان باجراء اشعة صوتية تسمى بالدوبلر(Doppler ) لمحاولة معرفة الاشخاص
الاكثر خطورة للاصابة بالشلل و القيام ببعض الاجراءات لمن حدوثه
و فيما يلي الأعراض العامة للمصابين بالانيميا المنجلية
.شحوب واصفرار البشرة والشفتين
الخمول و الشعور بالتعب والإرهاق لأقل جهد.
فقدان الشهية.
فقر الدم (اقل من 10 مليجرام)
تضخم الكبد والطحال نتيجة عجز نخاع العظام عن إنتاج كريات الدم الحمراء و نتيجة الى تشبع الطحال بالخلايا المنجلية و تكسرها فيه.
التأخر في النمو الجسماني كالطول والوزن .
تغيرات في عظام الجسم ومنها الجمجمة ، بروز الجبهة وعظام الوجنتين ، انخفاض عظام الأنف وبروز عظام الفك العلوي.
زيادة الالتهابات بشكل عام.
سرعة ضربات القلب لمحاولة تعويض نقص الهيموجلوبين وذلك بزيادة سرعة ضخ القلب للدم، كذلك قد تحدث مضاعفات قلبيه مع مرور الوقت.
اذا كثرة عدد مرات نقل الدم قد تحدث مضاعفات نتيجةلتراكم الحديد في أعضاء
مختلفة من الجسم أن لم يعطى المريض حقنة الدسفيرال والتي تؤدي إلى التخلص
من الحديد عن طريق البول وتتمثل هذه المضاعفات في تشحم الكبد ، اسوداد لون
الجلد، خلل هرموني (كمرض السكري او انخفاض في هرمون الغدة الدرقية او هرمون
الجار درقية او هرمونات الغدة النخامية .
مشاكل في القلب كتضخم عضلة القلب مع هبوط في القلب وعدم أنتظام في دقات القلب .
احتمال أصابه المريض بالأمراض المعدية نتيجة لنقل الدم بشكل مستمر ولكن
قلة هذه الامراض لان جميع مراكز التبرع بالدم تقوم بفحص تلقائي للكشف.
الانعكاسات النفسية السيئة لدى المريض نتيجة شعوره بالمرض الدائم والنقص الصحي مقارنة بأقرانه.
الاسبـــــــــــــاب :
الانيميا المنجلية مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض
أطفالهم.ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور
المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.ولكي تفهم هذا
الجانب العلمي أنصحك بالرجوع إلى صفحة أساسيات الوراثة لمعرفة ما هي
الخلية والنواة و الكروموسوم والمورث .وسوف أقوم بشرح مختصر عن هذا الكلمات
في سياق كلامي عن الهيموجلوبين والمورثات في هذه الصفحة.
تصنيع الهيموجلوبين:
يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع
العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها
البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم
(Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى بالجلوبين
(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع بيتا وفي هذه
الحالة يسمى هذا الهيموجلوبين بهيموجلوبين( أ ) أو (A ).
هذا الترابط العجيب بين الحديد و الهيم ،و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم.
كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي عبارة
عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل
الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات (الصبيغات)
،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين
يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات
موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض
الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة)
في المورثات التي تصنع مادة الجلوبين.
التمنجل و نقص الاكسجين :
الخلايا المنجلية موجودة باستمرار في العروق الدموية في الشخص المصاب بهذا
المرض.و تزيد شدتها عند ما ينقص الاكسجين في الدم .ان هذه الخلايا غير
طبيعية و لذلك هي قابلة للتكسر و التحلل.ويكثر التكسر في الطحال و لذلك
يكون متضخما في كثير من الاطفال خاصة خلال الاعوام الستة من العمر،ثم يقل
التضخم و ذلك لعدة اسباب و اهمها هو نتيجة لضمور الطحال نتيجة لكثرت
التجلطات و الانسدادات التي تحدث في الممراته الدموية .اذا كثرت تكسر الدم
يسب فقر دم و هو ايضا يرفع مستوى المادة الصفراء و لذلك فكثير من المصابين
تكون بشرتهم و عيونهم صفراء الون و هذا ما يعرف باليرقان.
هناك عوامل تزيد من حالة التمنجل ،و اهمها نقص الاكسجين(و ذلك عند الصعود
الى المرتفعات الشاهقة،الجفاف نتيجة لعدم تناول كمية كبيرة من السوائل ، و
نتيجة لحدوث الالتهابات المختلفة. اذا زادة حالة التمنجل في الدم او نقصة
السوائل في الجسم فان حركة الخلايا المنجلية في الدم تكون بطيئة و قد تشتد
سوء فينسد او يتجلط العرق الدموي او الشعيرة الدموية و التي تؤدي الى توقف
وصول الدم الى بعض الاعضاء و هذا يؤدي الى موت الخلايا و ضمورها.فاذا كان
العرق يغذي العظم حدثت الام مبرحة في تلك المنطقة و لو كان في الرئتين حدثة
حالات مشابة للالتهاب الرئوي و لو كانت في المخ لحدثة جلطة في المخ و ادت
الى شلل نصفي لا قدر الله.
المورثات و الجلوبين:
هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى
تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان
من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على
الكروموسومات،كما هو الحال في مادة الهيم.
ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم
11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11
الذي ورثة الإنسان من أمة.بينما يصنع مادة الألفا جلوبين من أربع
مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين
على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من
الأم.
الانيميا المنجلية هي ناتجة عن عطب في كلا الجينين الذين يصنعان مادة
البيتا جلوبين.و بالتحديد تختل ترتيب الاحماض النووية في فتؤدي الى وضع
الحمض الاميني فالين(Valine ) مكان الحمض الاميني المسمى بحمض الجلوتاميك(
Glutamic acid)
و الانيميا المنجلية لا تكون ناتجة عن خلل في الالفا جلوبين اطلاقا.و لكن
قد يحدث ان يوجد شخص مصاب بالانيميا المنجلية و الثلاسيميا من نوع الالفا
او البيتا في ان واحد.
الطفرة و البيتا جلوبين:
الحامل للانيميا المنجلية:
لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات البيتا جلوبين
فأن الخلية تنتج نوعين من البيتا جلوبين وذلك نظرا لوجود مورث أخر سليم
.مورث ينتج بيتا جلوبين طبيعية( يصّنع هيموجلوبين طبيعي) و مورث يتنج بيتا
جلوبين به طفرة (يصّنع منه هيموجلوبين منجلي).ويسمى الشخص في هذه الحالة
بالشخص الحامل للانيميا المنجلية. ويمكن أن يتعرف على هذه الحالة بأجراء
فحص للكريات الدم الحمراء والهيموجلوبين و اختبار التمنجل( Sickling test) و
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) و هي
متوفرة في الكثير من المستشفيات.
المصاب بالانيميا المنجلية:
أما لو حدثت الطفرة في كلا النسختان فان الخلية تنتج بيتا جلوبين غيرطبيعي
(و الذي بتالي يصنع هيموجلوبين منجلي ) ويمكن أن يتعرف على هذه الحالة
بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و اختبار التمنجل( sickling
test) و فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)
الوراثة و الانيميا المنجلية:
تنتقل الانيميا المنجلية من جيل الى اخر بما يعرف بالوراثة
المتنحية.ولمعرفة المزيد من هذا النمط يمكنك الرجوع الى صفحة الوراثة
المتنحية.و للوراثة المتنحية عدة خصائص و مميزات.فالمرض الذي ينتقل بهذه
الطريقة(كالانيميا المنجلية و الثلاسيميا)يصيب الذكور والاناث.كما انه
يشترط ان يكون كلا الوالدين حاملين للمرض،و ان هذه الاسرة المصابة يكون
فيها خليط من الاطفال المصابة و الحاملة للمرض و السليمة.و ان نسبة احتمال
الاصابة في كل مرة تحمل فيها المرأة تكون 25% و احتمال ان يولد طفل سليم
75% و بعض هؤلاء الاطفال السليمن يكونون حاملين للمرض مثل والديهم(بنسبة
60% تقريبا).
و اليك هذه الرسوم التي توضوح بشكل من التفصيل هذه النسب.الون الوردي يعني
الن الشخص سليم(كلا المورثين سليمين).اما الون الازرق الفاتح فيعني ان
الشخص مصاب بالانيميا المنجلية(كلا المورثين معطوبين).اما اذا كان الونين
الوردي و الازرق الفاتح موجودين مع بعض فان الشخص حامل للمرض(مورث سليم و
اخر معطوب)
الزوج و الزوجة سليمين :
لو تزوج زوجين كلاهما سليم(اي غير مصاب او حامل للأنيميا المنجلية) ،فانه
بمشيئة الله يكون جميع أطفال هذه الاسرة سليمة و غير مصابة بالانيميا
المنجلية (اي 100% سليم)
الزوج حامل للأنيميا المنجلية و الزوجة مصابة :
لو تزوج زوجين كلاهما حامل للأنيميا المنجلية فان أمام هذه العائلة أربع
احتمالات في كل مره تحمل فيه الزوجة باذن الله. .اثنان من هذه الأربعة
أطفال حاملين للمرض ( 4:2 أي 50% ) وثلاثة من هذه ألا ربعه أطفال مصابين
بالمرض (4:2 أي 50%) .
راجع مربع بينت للوراثة المتنحية لزوج مصاب و زوجة حاملة للمرض
راجع مربع بينت للوراثة المتنحية لزوجة مصابة و زوج حامل للمرض
الزوج سليم و الزوجة حاملة للأنيميا المنجلية :
لو تزوج زوجين احدهما حامل للانيميا المنجلية و الاخر سليم،فان أمام هذه
العائلة أربع احتمالات في كل مره تحمل فيه الزوجة باذن الله. .اثنان من هذه
الأربعة أطفال حاملين للمرض ( 4:2 أي 50% ) والاثنين الباقيين من هذه
الاربعه أطفال سليمين (4:2 أي 50%) .
الزوج سليم و الزوجة مصابة بالانيميا المنجلية :
لو تزوج زوجين احدهما مصاب بالانيميا المنجلية و الاخر سليم،فانه بمشيئة
الله يكون جميع اطفال هذه الاسرة حاملين للانيميا المنجلية (اي 100%
حاملين
الزوج و الزوجة مصابين بالانيميا المنجلية :
لو تزوج زوجين كلاهما مصاب بالانيميا المنجلية ،فانه بمشيئة الله يكون جميع
اطفال هذه الاسرة مصابة بالانيميا المنجلية (اي 100% مصاب)
الزوج و الزوجة حاملين للانيميا المنجلية:
لو تزوج زوجين كلاهما حامل للانيميا المنجلية فان أمام هذه العائلة أربع
احتمالات في كل مره تحمل فيه الزوجة باذن الله. الاحتمال الأول من هذه
الأربعة طفل سليم من المرض(1:4 أي 25% ) و اثنان من هذه ألا ربعه أطفال
حاملين للمرض (4:2 أي 50% ).و واحد من هذه الأربعة طفل مصاب بالمرض (4:1
أي 25% ). وبشكل أخر ففي كل مرة تحمل فيها الزوجة لديها 25% أن يولد لها
طفل مصاب و 75% أطفال غير مصابون ولكن بعضهم قد يكون حاملا للمرض(النسبة
تكون 2:3 أي 66 %) .
راجع مربع بينت للوراثة المتنحية للاب و الام حاملين للمرض
الزوج حامل للبيتا ثلاسيميا و الزوجة حاملة للانيميا المنجلية:
لو تزوج زوجين احدهما حامل للانيميا المنجلية و الاخر حامل للبيتا ثلاسيميا
فان أمام هذه العائلة أربع احتمالات في كل مره تحمل فيه الزوجة باذن
الله. الاحتمال الأول من هذه الأربعة طفل سليم من المرض(1:4 أي 25% ) و
الاحتمال الثاني طفل حامل للانيميا المنجلية (1:4 أي 25% ).و الاحتمال
الثالث هو طفل حامل للثلاسيميا (1:4 أي 25% ).و الاحتمال الرابع هو طفل
مصاب بالانيميا المنجلية و البيتا ثلاسيميا و يظهر المرض كانه انيميا
منجلية متوسطة الشدة (1:4 أي 25% ).
التشخيــــــص :
الهيموجلوبين وفقر الدم الناتج عن تكسر الدم:
الهيموجلوبين عبارة عن بروتين موجود في كريات الدم الحمراء وظيفته نقل
الأكسجين من الرئة إلى كافة خلايا الجسم ،وهو يتكون من الهيم (مادة الحديد +
صبغه) و الجلوبين (البروتين). وهناك أنواع مختلفة من بروتينات الجلوبين
المهمة منها الألفا ،البيتا ، الدلتا و الجاما جلوبين .ويتغير نوع
الهيموجلوبين في الجسم خلال الحمل إلى عدة شهور بعد الولادة نتيجة لتغير
نوع الجلوبين المنتج:
فعند الولادة يكون اغلب الهيموجلوبين(بنسبة تزيد عن 80%) من ما يعرف باسم
الهيموجلوبين الجنيني ( Hb F) ويستمر تواجده إلى أن تتم عملية إنتاج
الهيموجلوبين البالغين( Hb A) بشكل منتظم خلال الستة شهور الأولى من
العمر.,كما يوجد نسبة قليلة (1.5-3.5%) من هيموجلوبين يسمى بالهيموجلوبين
البالغين نوع 2 (( Hb A2) ).
المعدل الطبيعي للهيموجلوبين عند البالغين الذكور = 13-16 جرام/ 100 ملليليتر وعند البالغات الإناث = 12-15 جرام/ 100ملليليتر .
طريقة التشخيص
في العادة يكون لدى الشخص المصاب بالانيميا المنجلية اعراض لانخفاض
للهيموجلوبين( شحوب في البشرة)مع اصفرارا في البشرة(يرقان)و تضخم في الطحال
. و قد تنتفخ اليدين و القدمين ومع بكاء و تعب نتيجة لالم في العظام خاصة
في السنوات الاولى من العمر.و قد تكون حالة الطفل حرجة اذا كان مصحوب
بالتهاب بكتيري في الدم.ان و جود هذه الاعراض بحد ذاتها غير كافي لتشخيص
المرض و لذلك يجب اجراء ثلاثة تحاليل لتشخيص المرض:
:1-تحليل صورة الدم الكاملة( Comple Blood Count CBC):
فالشخص المصاب يكون مستوى الهيموجلوبين منخفض(بين 7 الى 10 مليغرامات لكل
100 مليلتر).و يكون حجم الكريات الحمراء و مستوى كريات الدم البيضاء و
الصفائح الدمويوة طبيعي.بينما الشخص الحامل للمرض فان هذا التحليل يكون
سليم،اي ان مستوى الهيموجلوبين غير منخفض.ولذلك هذا التحليل لا يفيد في
اكتشاف الشخص الحامل للمرض.
2- تحليل "تمنجل" الدم( sickling test) :
و هذا تحليل بسيط يتم عن طريق تعريض شريحة زجاجية عليها عينة من الدم الى
مادة مؤكسدة فظهر تحت المكبر الخلايا المنجلية بوضوح. وهذا التحليل من اهم
التحاليل التي يمكن اجرائها على عدد كبيرمن الناس لمعرفة من لدية خلايا
منجلية ام لا.وكل شخص الذي لديه خلايا منجلية بهذا التحليل قال له ان
تحليلة ايجابي المنجلي( Positive sickle test).و كل شخص لدية هذا التحليل
ايجابي يكون حامل للانيميا المنجلية او مصاب . ولذلك فهذا التحليل لا يفرق
بين الشخص الحامل للمرض و المصاب.ولذلك يجب اجراء التحليل رقم ثلاثة.
3- اختبار حركة الهيموجلوبين الكهربائية:
و هذا التحليل متوفر في الكثير من المستشفيات و هو يعطي قياس لنسبة
الهيموجلوبينات المجتلفة في الدم.و هم هيموجلوبين في حالة الشخص المصاب
بالانيميا المنجلية هو نسبة الهيموجلوبين المنجلي( HBs)فاذا كانت النسبة
اقل من 50% فالشخص حامل للانيميا المنجلية اما اذا كانت اعلى من هذه النسبة
فالشخص مصاب بالانيميا المنجلية. و اذا لم يوجد الهيموجلوبين فالشخص غير
حامل و لا مصاب بالانيميا المنجلية.
العـــــــلاج :
مرض الانيميا المنجلية من الامراض الزمنة التي لا يمكن الشفاء منها الا ان
يشاء الله.و لذلك من المهم الاهتمام بنصائح الاطباء و ارشاداتهم و
المتابعة المستمرة مع طبيب تثقون به .لقد تطور العلاج لهذا المرض في الفترة
الاخيرة فدواء الهيدروكسي يوريا و زراعة نخاع العظم و العلاج الجيني من
الامور التي لها مستقبل في مكافحة هذا المرض و التقليل من المشاكل الصحية
التي قد تصاحب هذا المرض .
من حسن الحظ ان العديد من المشاكل يمكن منعها او التقليل من شدتها.هناك
اجراءات لو اتخذت باذن الله تساعد المصابين بهذا المرض على ان يحيوا حياة
طبيعية و هنيئة .
نصائح عامة:
1- هناك أدوية معينة يجب أخذها بشكل منتظم ودقيق وعدم التوقف عن ذلك مهما
كنت الأسباب ، ومنها المضاد الحيوي ( بنسلين مثلاً ) وذلك عن طريق الفم
مرتين في اليوم حتى يصل عمر المريض إلى أكثر من 6-7 سنوات دواء حمض الفوليك
ا( Folic cid) وذلك حبة يومياً(5 مليجرام) باستمرار وذلكلزيادة الحاجة له
نتيجة لنشاط نخاع العظم لتعويض الانحلال المستمر لكرات الدم الحمراء .
2- يجب إعطاء الشخص المصاب بهذا المرض تطعيمات أخرى إضافية غير التطعيمات
المعتادة مثل تطعيم ضد جرثومة الألتهاب الرئوي نيموكوكيص( Pnumococus
vaccine ) وتطعيم ضد جرثومة هيموفيلس أنفلونزا (إHemophilus Influanza B)
وتطعيم ضد الجرثومة المسببة لالتهاب السحايا ـ الحمي الشوكية ميننجوكوكال (
meningococus) . وعند إتباع ما ذكر في الفقرتين 1و 2 فإن نسبة حماية الشخص
من الالتهابات البكتيرية القاتلة تكون عالية جداً بمشيئة الله .
3- يجب على الطبيب المعالج تدريب والدي المريض المصاب على معرفة طريقة قياس
حجم الطحال وتسجيل ذلك باستمرار وخاصة عند ملاحظة أي تغير مفاجئ في حالة
الطفل مثل شحوبه اللون ، اصفرار العينين بشكل أكبر عن المعتاد أو خمول وعدم
النشاط .
4- تناول غذاء صحي متوازن وبشكل جيد يحتوي على المجموعات الغذائية المختلفة .
5- تناول كمية من السوائل بشكل يويمي مستمر.و تزاد الكمية عند حدوث الاحمى او الاسهال او القيء او الم في العظام.
6- في فصل الشتاء ، يجب الاهتمام بتدفئة الجسم بشكل مستمر وخاصة أطراف الجسم ( اليدين والساقين ) .
7- عدم لبس ملابس ضيقة قد تضغط على الأوعية الدموية الدقيقة مما يؤدي إلى حدوث مضاعفات ( تحول الخلايا الحمراء إلى الشكل المنجلي ) .
8- الاهتمام بإعطاء الجسم قسطاً كافياً من الراحة وعدم مزاولة النشاطات بشكل كبير.
9- العناية بالأسنان بشكل مستمر وذلك بتنظيف الأسنان بشكل منتظم بعد كل
وجبة ومراجعة عيادة الأسنان بانتظام حيث أن أي تسوس في الأسنان قد يؤدي إلى
التهابات خطيرة .
10- ممارسة التمارين الرياضية بشكل معتدل وعدم الإفراط فيها .
11- تجنب تسلق الجبال العالية أو ركوب الطائرات غير مكيفة الضغط حيث أن
نسبة الأوكسجين قليلة مما قد يسبب الآلام في العظام وغيره من الجسم .
12- الحرص على المحافظة على المتابعة الدائمة للمواعيد بشكل منتظم ودقيق
وذلك في عبادة أمراض الدم وعند عدم التمكن من الحضور في الموعد المحدد يجب
الحصول على أقرب موعد ممكن والحرص على عمل التحاليل المطلوبة من الطبيب قبل
موعد العيادة .
13- إخبار المدرسة عن طبيعة مرض الطفل المصاب بفقر الدم المنجلي ولا مانع
من الاتصال بالطبيب من قبل إدارة المدرسة حتى يتمكن من تزويد المدرسة
بالمعلومات الكافية عن المرض وذلك حتى تكون المدرسة على إدراك كامل بالمرض
ومضاعفاته واكتشافها مبكراً .
14- المتابعة الطبية في المستشفى مع طبيب امراض الدم او طبيب اطفال متمرس في هذا المرض.
طرق الوقاية من حدوث نوبات انسداد للاوردة و اشعيرات الدموية(نوبات الالم):
منع الجفاف و ذلك باتناول كميات كبيرة من السؤال.
ينصح ان يتم و ضع علبة ماء (لتر و نصف )للاطفال اللذين يتجاوز عمرهم
الثالثة في الثلاجة و على الاهل متابعة و تشجيع الطفل على ان تناول هذه
الكمية خلال 24 ساعة.يمكن الاستعاضة عن الماء باي مادة سائلة كالعصير او
المشروبات الاخرى.عند حدوث اارتفاع في درجة الحرارة(حمى)يجب زيادة هذه
الكمية الى تقريبا 2 لتر او 2 و نصف ان امكن.عن حدوث اسهال فيجب اعطاء كوب
ماء (250 ميليلتر)عن كل مرة يسهل فيها الطفل.و يفضل ان يستبدل الماء في
حالة الاسهال بمحلول الجفاف المتوفر في الصيدليات او تحضيرة في المنزل اذا
تعذر الامر.يوجد في الصيدليات محاليل جفاف بطعم العصير يمكن استعمالة اذا
حدث صعوبة في اقناع الطفل على تناول السوائل.اذا حدث ارتفاع في درجة
الحرارة او اسهال و رفض الطفل تناول السوائل عليك بالتوجة الى
المستشفىلاعطاء شوائل عن طريق الوريد.
ان الجفاف و نقص السوائل تزيد من التمنجل في الدم والتي بالتالي تؤدي الى انسداد العروق ثم يتبعها الام مبرحة في العظام.
عليك الاحتياط بتناول الكثير من السوائل في الحالات التالية:
1-التعرق الشديد مع الجفاف (كالحمى)
2- الإجهاد و العب
3- الإلتهابات بجميع أنواعها خاصة المصوبة بارتفاع في درجة الحرارة.
4- الإسهال أو الاستفراغ الشديد المؤدي إلى الجفاف .
متى يجب الذهاب الى الطبيب :
يجب احضار الطفل الى الطبيب في الحال إذا كان مصابا بأحد من الاعراض التالية:
1- ارتفاع في درجة حرارة الجسم .
2- الألام في العظام او في الصدر يزيد في الشدة او لا يزول .
3-ألم شديد في البطن او انتفاخ مفاجئ وحاد في البطن .
4- شحوب مفاجئ في لون الوجه .
5- ضيق في التنفس او الم في الصدر
6- زيادة حادة في اصفرار الجسم وخاصة في العينين .
7- تضخم شديد ومفاجئ في الطحال .
8- صداع مفاجئ شديد أو تغير في مستوى الوعي ( الانتباه ) ،او تشنج
9- الآم مفاجئة شديدة في الجهاز التناسلي الذكري .
10-أسهال أو قيء
11-ءزيادة اصفرار العينين بشكل و اضح
12- ضعف مفاجئ في الاطراف
12- زغللة في العينين او ضعف مفاجئ في النظر.
علاج حالات الالم في العظام:
ان الوقاية من هذه النوبات هي العلاج الانجع و الاسهل.ولكن لو حدثة هذه
النوبات فحرص على ان يتلقى الطفل كفايتة من الادوية المسكنة للالم اضافة
الى الكثير من السوائل:
تناول الكثير من السوائل:
كما ذكرنا سابقا يجب ان تكون الكمية كبيرة و هي حوالي ضعف الكمية الذي يتناولها الطفل في العادة من دون ان يكون مريض.
الأدوية المسكنة للالم:
1- ان دواء خافظ الحرارة ايضا يخفف الالم اذا كان خفيف .
و جرعة هذا الدواء يختلف حسب وزنا لطفل.فالاطباء يعطون 10 مليجرامات من
البراسيتامول لكل كيلوجرام من وزن الطفل.ان معظم الادوية الخافظة للحرارة
عبارة عن مادة اسمها البراسيتامول( Paracetamol) . و يسوق هذا الدواء
باسماء(ماركات) مختلفة( الأادول ، البنادول، الفيفادول،التمبرا،التيلنول،و
غيرها).وكلها تحتوي نفس المادة من البراسيتامول.كما انه يمكن اعطاء الدواء
على شكل اقراص او شراب او تحميلة شرجية.و كلها لها نفس المفعول.يعطى
الدواء كل اربع الى ست ساعات ..الجرعة حسب العمر هي تقريبا 62.5 مليغرام
لاقل من 12 شهر،125 مليغرام لسن 1-4 سنوات،و 250 مليغرام لسن 4-10 سنوات،و
500 مليغرام 10-14 سنة،و 1000 مليغرام لمن هو فوق 15سنة.
ا2- ذا استمر الالم و الطفل يتناول البراسيتمول فانه يجب اعطاء دواء اقوى.
دواء الكودين فسفيت(Codeine phosphate ).و لا يمكن اخذه الى بوصفة من
الطبيب او في المستشفى.و يعطى بشكل منتظم كل 6-8 ساعات على حسب شدة الالم.و
الجرعة هي 1 او 2 مليجرام لكل كيلو جرام من وزن الطفل. و هو يفيد للالم
المتوسط الشدة،و ينصح ان يتناول مع البراسيتامول ليزيد المفعول. اذا لم
يتوفر دواء الكودين يمكن تجربة دواء ديكلوفيناك ( Diclofenac) , و يسمى
تجاريا بالفولتارين (Voltaren).و الجرعة هي 1 مليجرام لكل كيلو جرام من و
زن الطفل. و يمكن ان يعطى 3 مرات في اليوم. و يوجد منه اقراص او تحميلات
شرجية او حقن عضلية او و ريدية.
3- اذا لم تفد الادوية اعلاها او كان الالم شديد فيجب اعطاء دواء
المورفين(morphine ) او بيثيدين ( Pethidine)عن طريق الوريد اما عن طريق
جرعات متفرقة او مستمرة .في المستشفى و يجد طريقة تسمح للطفل الكبير ان
يتحكم في الجرعة التي يتناولها عن طريق ضغاط تحكم في الممغذي الذي عن طريقة
يمر الدواء.
وسائل علاج الأنيميا المنجلية: تحت الانشاء
العلاج بعقار هيدروكسي يوريا (Hydroxyurea)
في شهر يناير من عام 1995 أوقف الباحثون المشتركون في دراسة لمعرفة هل"
الهيدروسكي يوريا مفيد لتقليل نوبات الألم للمصابين بمرض الأنيميا
المنجلية" بحثهم بعد أن أظهرت النتائج إن الهيدروكسي يويا يقلل نوبات الألم
و عدد مرات الدخول إلى المستشفى بنسبة 50 % مقارنة بمجموعة أخرى من المرضى
الذين أعطوا دواء غير فعال(Placebo) للمقارنة .لقد أوقفوا البحث و الذي
اشترك فيه 21 مركز طبي قبل أربعة اشهر من نهايته أن يكملوه و ذلك للنتائج
الغير متوقعة في نجاح الدواء في التقليل من عدد نوبات الألم و يعرف ذلك
البحث بين الاطباء ببحث الهيروكسي يوريا ذو المراكز المتعددة (Multicenter
Study of Hydroxyurea (MSH Trial)).
اجري ذلك البحث على البالغين و الذين أعمارهم فوق الثامنة عشرة. و قد
وافقة الوكالة الأمريكية للغذاء و الدواء (FDA)على التصريح باستخدام هذا
الدواء لعلاج الأنيميا المنجلية.
أما بنسبة للأطفال فقد اجري بحث على 50 طفل لمن أعمارهم بين 5 إلى 15 سنة و
قد اثبت هذا الدواء فائدة في تقليل عدد مرات نوبات الألم كما هو الحال في
الكبار مع عدم ظهور أعراض جانبية لهذا الدواء على النمو أو على أعضاء الجسم
خلال مرور سنة من تناول هذا الدواء.و لكن الوكالة الأمريكية للغذاء و
الدواء لم تصرح إلى الآن باستعمال هذا الدواء للأطفال حتى تنتهي بقية
الأبحاث المتعلقة بالآثار الجانية لهذا الدواء. و قد وصلت هذه الأبحاث إلى
المستوى الثالث و يتوقع أن تظهر النتائج في خلال السنوات القادمة.
كما اجري بحث أخر على الأطفال الذين تتراوح أعمارهم بين الستة اشهر و 24
شهر و أظهرت تلك الدراسة أن هؤلاء الأطفال يتقبلون الدواء بشكل جيد.
و يعتقد الكثير من الأطباء أن هذا الدواء قد يمنع حدوث تلف في أعضاء الجسم
المتفرقة كالقلب و الكبد و و الغدد الصماء و الجهاز العصبي و لذلك فهم
يحاولون أن يقوموا بالمزيد من الأبحاث لإثبات هذه الاعتقادات.
كان و مازال دواء الهيدروكسي يوريا يستعمل في علاج أمراض الدم المرتبطة
بنشاط فمفرط و غير فعال في نخاع العظم كمرض البولي سيثيميا (polycythemia
vera) و الأورام التي لها علاقة بالدم و العقد اللمفاوية.و هذا الدواء لا
يستخدم للشفاء من الأنيميا المنجلية بل هو فقط يساعد في تقليل نوبات الألم و
التي تمثل المشكلة الكبر للمصابين بهذا المرض. لا يعرف الأطباء بشكل دقيق
كيف يعمل هذا الدواء في تقليل تلك النوبات و أن كان بعض العلماء يعتقدون أن
ذلك يحدث عن طريق رفع مستوى هيموجلوبين الجنين ( Hb F fetal hemoglobin) و
هذا بالتالي يقلل من تلاصق كريات الدم الحمراء المنجلية مع بعضها البعض و
لا يحدث انسداد في الشعيرات الدموية.كما انه لوحظ أن هذا الدواء يرفع هرمون
الاريثروبيوتين و الذي تفرزه الكلية لتنشيط و حث نخاع العظم لتصنيع
المزيد من كريات الدم الحمراء.كما إن هذا الهرمون أيضا يرفع مستوى
الهيموجلوبين الجنين ، و هذا ما جعل الأطباء يحاولون استخدام هذا
الهرمون(عن طريق إعطائه عن طريق الوريد) مع الهيدروكسي يوريا لعلاج هذا
المرض.
قد لا يفيد هذا الدواء جميع المصابين بالأنيميا المنجلية حيث أن هذا
الدواء استعمل لمن يعانون من نوبات الم متكررة و شديدة.و لذلك قد لا يستعمل
لمن حالتهم اقل شدة و ذلك لان هذا الدواء يصنف على انه من الأدوية التي
تستخدم للأورام و السرطان ( Cytotoxic Drug ) . كما أن الفائدة من الدواء
لا تظهر إلا بعد استعماله لفترة من الزمن لا تقل عن شهر بأية حال و على
المريض أن يستمر في تناوله إلى أن يقرر الطبيب غير ذلك.و
و يعطى الهيدروكسي يوريا عن طريق الفم و يبدأ بجرعة قليلة ثم تزاد الجرعة
تدريجيا إلى أن تظهر الفائدة او تظهر الأعراض الجانبية و التي تتمثل في
انخفاض كريات الدم البيضاء او الصفائح،و في هذا الحال يوقف الدواء مؤقتا ثم
يعاد البدء في إعطائه مرة أخرى بجرعة اقل.
العلاج بهرمون الاريثروبيوتين( erythropoietin )
العلاج باالجيني (Gene Therapy)
نقل دم
ينقل الدم عن الزوم و عندما ينخفض مستوى الهيموجلوبين عن 7 مليغرام لكل لتر
. و قد يستدعي نقل الدم لبعض الحالات خاصة قبل اجراء العمليات الجراحية او
عند حدوث انسداد في العروق الدموية في الرئتين . كما يقوم الطبيب بنقل
الدم بشكل دوري و مستمر كل 3-4 اسابيع لك الاشخاص الذين سبق ان اصيبوا بشلل
نصفي ناتجة عن جلطة في المخ.
العلاج بالديسفسرال
يترسب الحديد في جسم المريض تدريجيا مما يؤدى إلى تعطيل وظائف الخلايا
وبالتالي إلى موتها وفقدان وظائفها، كما يؤدى إلى الكثير من اختلال الوظائف
الهرمونية و القلبية و الكبدية و الجلدية. لذا يجب التخلص من الحديد
الزائد (اكثر من 1000 مج\مل). الديسفرال عبارة عن مادة ترتبط مع الحديد في
الجسم وتخرج مرتبطة بالحديد لخارج الجسم عن طريق البول، ومن هنا تأتى أهميه
الديسفرال الذي يحقن عن طريق الجلد لفتره8-10 ساعات يوميا او عن طريق
الوريد،والعضل
إزالة الطحال
للطحال دور هام في التخلص من كريات الدم الميتة نتيجة مشاركة الطحال في
تعويض الجسم عن عجز نخاع العظم واستمرار تراكم الحديد بصوره كبيره يبدأ
بعدها الطحال بتدمير كريات الدم الحمراء والبيضاء والصفائح الدموية ،تتفاقم
حدة فقر الدم وتتناقص مناعة ومقاومة المريض ويصبح عرضة للنزف، ويتم
استئصال الطحال عندما تظهر المؤشرات التالية:
عندما يحتاج المريض إلى نقل كميات من الدم تزيد بمقدار واحد ونصف عن الكميه الكافية.
نقص حاد في كريات الدم البيضاء أو الصفائح الدموية نتيجة تدميرها في الطحال
التضخم الكبير والمتزايد للطحال.
زرع نخاع العظم (Bone Marrow Transplant)
هذه العملية تعتمد على وجود متبرع يفضل أن يكون من أشقاء او شقيقات المريض
حيث يجب أجراء الفحوصات التي تؤكد من وجود التطابق النسيجي والخلوي (100%)
بين المتبرع والمريض،وتزداد فرص نجاح العملية للذين لا يعانون من ترسب
الحديد أو تضخم الكبد والطحال.
إن رفض الانسجه عادة ما يكون اقل نسبة في الأطفال عنه في الكبار فذلك لا
يحدث مباشرة بعد العملية إنما قد يحتاج إلى 3-5 سنوات يبقى فيها خطر
الرفض،فالمريض بحاجة إلى متابعة دقيقه للغاية.
ويتم فبل زرع نخاع العظام التخلص من نخاع المريض بتناوله الأدوية تدمر
خلايا النخاع وعلى مدى 6 أيام يشعر فيها بالوهن والضعف وتحت التعقيم التام
يتم شفط النخاع بواسطة حقنه خاصة وتحت التخدير العام وتستغرق هذه العملية
حوالي نصف الساعة وتستكمل العملية بإعادة حقن نخاع العظام السليم إلى
المريض فيما يشبه عملية نقل الدم فيجد النخاع الجديد تجويف نخاع المصاب.
والمتبرع عادة لا يعاني من أي مخاطر وجسمه قادر على تعويض الكميه التي تم
شفطها من نخاع العظام ولا يحتاج للبقاء في المستشفى اكثر من يوم واحد فقط.
الجديد في علاج الانيميا المنجلية:
بدأت بعض المراكز في إجراء عمليات نقل وزراعة نخاع العظم للجنين المصاب
داخل رحم أمه بدل إجهاضه وتتميز هذه العملية بندرة رفض جسم المريض للنخاع
المزروع.
طرق الوقاية:
فحوصات قبل الولادة
إذا كان كلا الوالدان يحملان الانيميا المنجلية فمن الممكن أن يتم فحص
الجنين للتأكد من سلامته من الاصابة بالانيميا المنجلية خلال الأسبوع
10-11 من الحمل. ويتم الفحص بأخذ عينه من المشيمة ( Chorionic Villus
)وتحليلاها بواسطة ال DNA لمعرفة إذا ما كان الجنين مصاب أم لا.،وتظهر
نتيجة التحليل خلال 3-4 أسابيع.وكما هو الحل في جميع الفحوصات الطبية فقد
يكون هناك بعض المضاعفات واهما احتمال الإجهاض من جراء سحب العينه 1%لثانية
هي عن طريق سحب من السائل الأمينوسي والموجود حول الجنين.وتأخذ هذه العينة
في الأسبوع 14 -16 من الحمل واحتمال الإجهاض في هذه الحالة حوالي نصف في
المائة 0.5%
فحوصات قبل الزواج :
يمكن الكشف على أي شخص لمعرفة إذا ما كان حامل للمرض و بطريقة سهلة و ميسرة
و متوفرة في معظم المختبرات الطبية .و هو تحليل رخيص في قيمته المادة لكنه
غالي في قيمته العلمية. ولا يتوقع أن يكلف هذا التحليل أكثر من 15
دولار.و ينصح بإجراء هذا التحليل لكل من له أصول عرقية بمنطقة الخليج
العربي و جنوب السعودية و اليمن و المناطق الشمالية الغربية من السعودية. و
إن كنا نفضل أن يجرى هذا التحليل إضافة إلى تحاليل المتعلقة بالثلاسيميا
على جميع من يريد الزواج بغض النظر عن أصولة العرقية و ذلك لرخص قيمته و
توفره .
الانيميا المنجلية هي احد أنواع فقر الدم.وهي تصيب كريات الدم الحمراء.و هي
من اشهر أمراض الدم الوراثية الانحلالية- التي تسبب تكسر كريات الدم
الحمراء-و هي اكثرها شيوعاً على مستوى العالم بشكل عام و في افريقيا و
الشرق الاوسط بشكل خاص . و هناك عدة اسماء لهذا المرض ،فاضافة الى الانيميا
المنجلية فانه يطلق عليها ايضا فقر الدم المنجلي او مرض المنجلية.وايا كان
الاسم فالمرض واحد و ليس هناك فروق بينها.
و كلمة المنجلية مأخوذه من المنجل(الذي يحصد به النبات و في بعض المناطق
يطلق علية المحش)و ذلك لان كريات الدم الحمراءتحت المجهر تأخذ شكل مقوس
كالمنجل او الهلال .وكلمة انيميا تعني فقر دم .و ينتشر المرض في عدة دول
افريقية و اسيوية.وهو تقريبا موجود في جميع الدول العربية .أما في بعض
مناطق من الخليج العربي (كمنطقة الاحساء و القطيف و جازان في
السعودية)فيحمل كل 4 افراد من بين 20 فرد هذا المرض، أي نسبه حاملي صفة
المرضحوالي 20 % ،و هي نسبه لا يمكن تجاهلها حيث أنه في كل مرة تحمل فيه
امرأة حاملة للمرض ومتزوجة من رجل أيضا حامل للمرض فإن احتمال إصابة الجنين
تصل إلى 25% وان اكثر من 60% من أطفال هذه الأسرة السليمين أيضاً حاملين
للمرض. وتكمن مشكلة المرض في انتاج نخاع العظم لكريات دم حمراء - التي تنقل
الغذاء والأكسجين إلى مختلف أنحاء الجسم – غير طبيعية نتيجة لخلل في تكوين
الهيموجلوبين( خضاب الدم) .وهذه الخلايا غير الطبيعية تأخذ شكل المنجل و
هي قابلة الى التكسر وتتحلل بعد فتره قصيرة من إنتاجها و قد تعيق مرور
الدم خلال الشعيرات الدموية، و قد تسد عروق الدم فتسبب الام مبرحه في
اجزاء مختلفة من الجسم خاصة في العظام خاصة عظام الاطرف و الظهر.و قد تسد
كريات الدم الحمراء المنجلية اي عرق من العروق الدموية في الرئتين او في
البطن او حتى في المخ و قد تسبب مضاعفات خطيرة اضافة الى الألام المببرحه
التي يعاني منها الشخص المصاب. .
ينتشر المرض في عدة مناطق من العالم ولكنها تكثر في المناطق التالية:
* افريقيا بشكل عام.
* منطقة الخليج العربي و في اليمن و جنوب شرق السعودية.
* منطقة الشرق الأوسط وتشمل إيران العراق سوريا الأردن وفلسطين.
* شبه القارة الهندية.
* جنوب شرق أسيا .
* المنطقة الكاريبية في امريكا الوسطى
الحامل للانيميا المنجلية (Sickle cell trait) :
يكون الشخص حامل للانيميا المنجلية اذا وجود مورث سليم لسلسة البروتين بيتا
و أخر معطوب،ولا يعاني حامل المرض في العادة من أي مشكلة صحية. و يمكن
التعرف على الشخص الحامل عن طريق فحص بسيط للدم.
المصاب بالانيميا المنجلية:
هو شخص لديه عطب في كلا الجينين المصنعين لسلسة البروتين بيتا ( Beta
Globin chain).و عند الولادة يكون الطفل سليم و لكن عند بلوغ الشهر السادس
او اكثر من العمر يبدأ ظهور علامات المرض و التي في العادة تكون مصحوبة
بانخفاض بنسبة الهيموجلوبين(فقر دم)مع شحوب في البشرة و انتفاخ في اليدين و
القدمين و بكاء ناتج عن الام في العظام.و قد تكون حالة الطفل حرجة اذا كان
مصحوب بالتهاب بكتيري في الدم.و في العادة يتراوح مستوى الهيموجلوبين
حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان .
ويحتاج الطفال لنقل الدم اذا انخفض الهيموجلوبين اقل من 7 غرامات او كانت .
مصاب الأنيميا المنجلية و بالثلاسيميا
في هذه الحالة يصاب المريض بمرضين في أن واحد.فيكون الشخص أما لدية مرض
الأنيميا المنجلية ومرض الثلاسيميا معا أو يكون حاملا للأنيميا المنجلية مع
مرض الثلاسيميا.وقد تتراوح شدة المرض وانقص الهيموجلوبين على حسب شدة
الإصابة في مورث البيتا او الالفا جلوبين.و فيالعادة تكون الاصابة في
الشخاص المصابين بالانيميا المنجلية و البيتا ثلاسيميا اقل شدة ممن يصاب
بالانيميا المنجلية لوحدها و ذلك حدوث الثلاسيميا يقلل من الكمية المنتجة
من الهيموجلوبين المنجلي و الذي يسبب مشاكل كثيرة و يسد الشعيرات الدموية.
المورث (الجين) المسؤول عن إنتاج مادة البيتا جلوبين: يسمى بمورث البيتا
جلوبين يوجد على الكروموسوم 11. و كل انسان لديه نسختان من هذا المورث
،نسخة تاتي من الاب و الاخرى من الام
الأعــــــراض :
لا تظهر اي اعراض على الشخص الحامل للانيميا المنجلية و قد تكتشف
بالمصادفة عند إجراء اختبار التمنجل والذي يجرى في العادة قبل اجراء عملية
جراحية حيث يجرى بشكل روتيني قبل العملية في كثير من البلدان.
اما بنسبة للمصاب الانيميا المنجلية فالاعراض تختلف على حسب سن المريض:
قبل الشهر السادس من العمر:
لا تظهراي اعراض خلال الاشهر الاولى من العمر حتى في الشخص المصاب
بالانيميا المنجلية.وهذا لان نخاع العظم(المصنع الذي ينتج كريات الدم
الحمراء)لا يبدأ في صناعة الهيموجلوبين الذي يحتاج الى البيتا جلوبين الى
بعد مضي عدة اشهر من الولادة.و كما شرحنا في قسم الاسباب فان البيتا جلوبين
تدخل في تكوين الهيموجلوبين S .
بعد الشهر السادس و خلال السنتين الاوليين من العمر:
من اهم الاشكال التي يظهر فيها المرض في هذا السن ما يعرف مرض اليدين
والقدمين.حيث تنتفخ اليدين و القدمين مصحوب بالم و بكاء .و قد يتركز التورم
في الاصابع . كما يكون بشرة الطفل شاحبة نتيجة لوجود فقر الدم.
في بعض الاحيان تكون هذه الحالة مصحوبة بالتهاب فيروسي او بكتيري في
الدم.وقد يكون هذا الالتهاب شديد و قد يستدعي ان يدخل الطفل العناية
المركزة.
بعدها تبدأ حدوث النوبات المتكررة من الالم و الناتج لانسداد العروق
الدموية المغذية للعظام خاصة عظام الاطراف و العمود الظهري.تختلف شدة هذه
النوبات و عدد مرات تكرارها من شخص الى اخر و يبدوا ان هناك نوعان من
الانيميا المنجلية .نوع يكون شديد و يتركز هذا في المنطقة الجنوبية الغربية
من السعودية (جازان و ماحولها من المناطق)و نوع اقل شدة في الحالات التي
يكون مصدرها من المنطقة الشرقية من السعودية.اتفاوت الشدة غير معروف السبب
بشكل كامل.و لكن هناك تكهنات بان المرض يكون قل خطورة كلما كانت نسبة
الهيموجلوبين F عليه.و قد يكون مستوى الهيموجلوبين F في الحالات التي في
المنطقة الشرقية من السعودية اعلى من الحالات التي يكون مرجعها المنطقة
الجنوبية من السعودية.هناك رأي اخر و ذلك يرجع الى الاصول الوراثية للاشخاص
المصابين بهذا المرض.فالمرض الذي في المنطقة الجنوبية يسمى بالبينين (
Benin) ويله اصول افريقية.بينما الذي في المنطقة الشرقية فيعرف بالسعودي
الهندي و ذلك لتشابهه للحالات التي في الهند.
خلا ل سنتين الى العاشرة سنوات من العمر:
في معظم الوقت تكون البشرة شاحبة نتيجة لوجود فقر الدم حيث يتراوح مستوى الهيموجلوبين بين 7-10 مليغرام.
اصفرار البشرة و العينين نتيجة لتكسر الدم و ارتفاع المادة الصفراء في الدم(يرقان)
اقد تكون البنية نحيفة ،مع تعب و ارهاق عند القيام باعمال فيها جهد عالي.
نوبات الالم.و سوف نفصل في هذه النوبات في اخر هذه الصفحة .
زيادة القابلية للاصابة بالالتهابات خاصة الالتهابات البكتيرية (خاص
Hemophilus Infeluanza و Pnueomococus ) وي مكن الوقاية منها باخذ القاح
الواقي منها(تطعيم).
تكثر الالتهابات افي الجهاز النفسي، و في بعض الاحيان يحدث التهاب في العظم
. و قد يلتبس او يصعب على الاطباء في هذه الحالة التمييز بين نوبة الالم
المتكرة الحدوث وبين التهاب العظم .وهذا مشهور بين الاطباء.
بعد سن العاشرة من العمر:
ان اهم ما يميز هذا العمر هو زيادة احتمال اصابة الرئتين بتجلطات نتيجة
لانسددا العروق الدموية بالخلايا المنجلية.و تظهر هذه الحالات على شكل الم
في الصدر مع كحة جافة و نقص في الاكسجين. و قد تلتبس او يصعب على الاطباء
التفرقه بينها و بين الالتهاب الرئوي.
شحوب مصحوب ببشرة داكنة مع بروز في عظام الجبهة والوجنتين والفك العلوي.مع
ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال
مشاكل صحية قد تحدث في اي عمر:
نوبات الم
التهابات متكررة
تقرح في الساقين يصعب شفائها
حصوات في المراراة.
انتصاب مؤلم عند الذكور (Priapism) .
فقر الدم الناتج عن نقص حمض الفوليك. و قد يظهر هذا على شكل انخفاض في
نسبة الهيموجلوبين مع كبر في حجم الخلايا الحمراء . وقد يتطور و تبداء
الخلايا البيضاء في الانخفاض لو كان النقص شديد. و الاطباء يعطون جميع
المصابين بالانيميا المنجلية حمض الفوليك بشكل يومي و ذلك لزيادة الحاجة
لهذا الفيتامين المهم في تكوين و صنع خلايا الدم خاصة الخلايا الحمراء.
حدوث شلل نصفي نتيجة لاتسداد احد العروق في المخ.يقوم الكثير من الاطباء
الان باجراء اشعة صوتية تسمى بالدوبلر(Doppler ) لمحاولة معرفة الاشخاص
الاكثر خطورة للاصابة بالشلل و القيام ببعض الاجراءات لمن حدوثه
و فيما يلي الأعراض العامة للمصابين بالانيميا المنجلية
.شحوب واصفرار البشرة والشفتين
الخمول و الشعور بالتعب والإرهاق لأقل جهد.
فقدان الشهية.
فقر الدم (اقل من 10 مليجرام)
تضخم الكبد والطحال نتيجة عجز نخاع العظام عن إنتاج كريات الدم الحمراء و نتيجة الى تشبع الطحال بالخلايا المنجلية و تكسرها فيه.
التأخر في النمو الجسماني كالطول والوزن .
تغيرات في عظام الجسم ومنها الجمجمة ، بروز الجبهة وعظام الوجنتين ، انخفاض عظام الأنف وبروز عظام الفك العلوي.
زيادة الالتهابات بشكل عام.
سرعة ضربات القلب لمحاولة تعويض نقص الهيموجلوبين وذلك بزيادة سرعة ضخ القلب للدم، كذلك قد تحدث مضاعفات قلبيه مع مرور الوقت.
اذا كثرة عدد مرات نقل الدم قد تحدث مضاعفات نتيجةلتراكم الحديد في أعضاء
مختلفة من الجسم أن لم يعطى المريض حقنة الدسفيرال والتي تؤدي إلى التخلص
من الحديد عن طريق البول وتتمثل هذه المضاعفات في تشحم الكبد ، اسوداد لون
الجلد، خلل هرموني (كمرض السكري او انخفاض في هرمون الغدة الدرقية او هرمون
الجار درقية او هرمونات الغدة النخامية .
مشاكل في القلب كتضخم عضلة القلب مع هبوط في القلب وعدم أنتظام في دقات القلب .
احتمال أصابه المريض بالأمراض المعدية نتيجة لنقل الدم بشكل مستمر ولكن
قلة هذه الامراض لان جميع مراكز التبرع بالدم تقوم بفحص تلقائي للكشف.
الانعكاسات النفسية السيئة لدى المريض نتيجة شعوره بالمرض الدائم والنقص الصحي مقارنة بأقرانه.
الاسبـــــــــــــاب :
الانيميا المنجلية مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض
أطفالهم.ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور
المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.ولكي تفهم هذا
الجانب العلمي أنصحك بالرجوع إلى صفحة أساسيات الوراثة لمعرفة ما هي
الخلية والنواة و الكروموسوم والمورث .وسوف أقوم بشرح مختصر عن هذا الكلمات
في سياق كلامي عن الهيموجلوبين والمورثات في هذه الصفحة.
تصنيع الهيموجلوبين:
يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع
العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها
البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم
(Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى بالجلوبين
(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع بيتا وفي هذه
الحالة يسمى هذا الهيموجلوبين بهيموجلوبين( أ ) أو (A ).
هذا الترابط العجيب بين الحديد و الهيم ،و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم.
كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي عبارة
عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل
الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات (الصبيغات)
،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين
يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات
موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض
الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة)
في المورثات التي تصنع مادة الجلوبين.
التمنجل و نقص الاكسجين :
الخلايا المنجلية موجودة باستمرار في العروق الدموية في الشخص المصاب بهذا
المرض.و تزيد شدتها عند ما ينقص الاكسجين في الدم .ان هذه الخلايا غير
طبيعية و لذلك هي قابلة للتكسر و التحلل.ويكثر التكسر في الطحال و لذلك
يكون متضخما في كثير من الاطفال خاصة خلال الاعوام الستة من العمر،ثم يقل
التضخم و ذلك لعدة اسباب و اهمها هو نتيجة لضمور الطحال نتيجة لكثرت
التجلطات و الانسدادات التي تحدث في الممراته الدموية .اذا كثرت تكسر الدم
يسب فقر دم و هو ايضا يرفع مستوى المادة الصفراء و لذلك فكثير من المصابين
تكون بشرتهم و عيونهم صفراء الون و هذا ما يعرف باليرقان.
هناك عوامل تزيد من حالة التمنجل ،و اهمها نقص الاكسجين(و ذلك عند الصعود
الى المرتفعات الشاهقة،الجفاف نتيجة لعدم تناول كمية كبيرة من السوائل ، و
نتيجة لحدوث الالتهابات المختلفة. اذا زادة حالة التمنجل في الدم او نقصة
السوائل في الجسم فان حركة الخلايا المنجلية في الدم تكون بطيئة و قد تشتد
سوء فينسد او يتجلط العرق الدموي او الشعيرة الدموية و التي تؤدي الى توقف
وصول الدم الى بعض الاعضاء و هذا يؤدي الى موت الخلايا و ضمورها.فاذا كان
العرق يغذي العظم حدثت الام مبرحة في تلك المنطقة و لو كان في الرئتين حدثة
حالات مشابة للالتهاب الرئوي و لو كانت في المخ لحدثة جلطة في المخ و ادت
الى شلل نصفي لا قدر الله.
المورثات و الجلوبين:
هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى
تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان
من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على
الكروموسومات،كما هو الحال في مادة الهيم.
ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم
11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11
الذي ورثة الإنسان من أمة.بينما يصنع مادة الألفا جلوبين من أربع
مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين
على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من
الأم.
الانيميا المنجلية هي ناتجة عن عطب في كلا الجينين الذين يصنعان مادة
البيتا جلوبين.و بالتحديد تختل ترتيب الاحماض النووية في فتؤدي الى وضع
الحمض الاميني فالين(Valine ) مكان الحمض الاميني المسمى بحمض الجلوتاميك(
Glutamic acid)
و الانيميا المنجلية لا تكون ناتجة عن خلل في الالفا جلوبين اطلاقا.و لكن
قد يحدث ان يوجد شخص مصاب بالانيميا المنجلية و الثلاسيميا من نوع الالفا
او البيتا في ان واحد.
الطفرة و البيتا جلوبين:
الحامل للانيميا المنجلية:
لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات البيتا جلوبين
فأن الخلية تنتج نوعين من البيتا جلوبين وذلك نظرا لوجود مورث أخر سليم
.مورث ينتج بيتا جلوبين طبيعية( يصّنع هيموجلوبين طبيعي) و مورث يتنج بيتا
جلوبين به طفرة (يصّنع منه هيموجلوبين منجلي).ويسمى الشخص في هذه الحالة
بالشخص الحامل للانيميا المنجلية. ويمكن أن يتعرف على هذه الحالة بأجراء
فحص للكريات الدم الحمراء والهيموجلوبين و اختبار التمنجل( Sickling test) و
فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis) و هي
متوفرة في الكثير من المستشفيات.
المصاب بالانيميا المنجلية:
أما لو حدثت الطفرة في كلا النسختان فان الخلية تنتج بيتا جلوبين غيرطبيعي
(و الذي بتالي يصنع هيموجلوبين منجلي ) ويمكن أن يتعرف على هذه الحالة
بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و اختبار التمنجل( sickling
test) و فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)
الوراثة و الانيميا المنجلية:
تنتقل الانيميا المنجلية من جيل الى اخر بما يعرف بالوراثة
المتنحية.ولمعرفة المزيد من هذا النمط يمكنك الرجوع الى صفحة الوراثة
المتنحية.و للوراثة المتنحية عدة خصائص و مميزات.فالمرض الذي ينتقل بهذه
الطريقة(كالانيميا المنجلية و الثلاسيميا)يصيب الذكور والاناث.كما انه
يشترط ان يكون كلا الوالدين حاملين للمرض،و ان هذه الاسرة المصابة يكون
فيها خليط من الاطفال المصابة و الحاملة للمرض و السليمة.و ان نسبة احتمال
الاصابة في كل مرة تحمل فيها المرأة تكون 25% و احتمال ان يولد طفل سليم
75% و بعض هؤلاء الاطفال السليمن يكونون حاملين للمرض مثل والديهم(بنسبة
60% تقريبا).
و اليك هذه الرسوم التي توضوح بشكل من التفصيل هذه النسب.الون الوردي يعني
الن الشخص سليم(كلا المورثين سليمين).اما الون الازرق الفاتح فيعني ان
الشخص مصاب بالانيميا المنجلية(كلا المورثين معطوبين).اما اذا كان الونين
الوردي و الازرق الفاتح موجودين مع بعض فان الشخص حامل للمرض(مورث سليم و
اخر معطوب)
الزوج و الزوجة سليمين :
لو تزوج زوجين كلاهما سليم(اي غير مصاب او حامل للأنيميا المنجلية) ،فانه
بمشيئة الله يكون جميع أطفال هذه الاسرة سليمة و غير مصابة بالانيميا
المنجلية (اي 100% سليم)
الزوج حامل للأنيميا المنجلية و الزوجة مصابة :
لو تزوج زوجين كلاهما حامل للأنيميا المنجلية فان أمام هذه العائلة أربع
احتمالات في كل مره تحمل فيه الزوجة باذن الله. .اثنان من هذه الأربعة
أطفال حاملين للمرض ( 4:2 أي 50% ) وثلاثة من هذه ألا ربعه أطفال مصابين
بالمرض (4:2 أي 50%) .
راجع مربع بينت للوراثة المتنحية لزوج مصاب و زوجة حاملة للمرض
راجع مربع بينت للوراثة المتنحية لزوجة مصابة و زوج حامل للمرض
الزوج سليم و الزوجة حاملة للأنيميا المنجلية :
لو تزوج زوجين احدهما حامل للانيميا المنجلية و الاخر سليم،فان أمام هذه
العائلة أربع احتمالات في كل مره تحمل فيه الزوجة باذن الله. .اثنان من هذه
الأربعة أطفال حاملين للمرض ( 4:2 أي 50% ) والاثنين الباقيين من هذه
الاربعه أطفال سليمين (4:2 أي 50%) .
الزوج سليم و الزوجة مصابة بالانيميا المنجلية :
لو تزوج زوجين احدهما مصاب بالانيميا المنجلية و الاخر سليم،فانه بمشيئة
الله يكون جميع اطفال هذه الاسرة حاملين للانيميا المنجلية (اي 100%
حاملين
الزوج و الزوجة مصابين بالانيميا المنجلية :
لو تزوج زوجين كلاهما مصاب بالانيميا المنجلية ،فانه بمشيئة الله يكون جميع
اطفال هذه الاسرة مصابة بالانيميا المنجلية (اي 100% مصاب)
الزوج و الزوجة حاملين للانيميا المنجلية:
لو تزوج زوجين كلاهما حامل للانيميا المنجلية فان أمام هذه العائلة أربع
احتمالات في كل مره تحمل فيه الزوجة باذن الله. الاحتمال الأول من هذه
الأربعة طفل سليم من المرض(1:4 أي 25% ) و اثنان من هذه ألا ربعه أطفال
حاملين للمرض (4:2 أي 50% ).و واحد من هذه الأربعة طفل مصاب بالمرض (4:1
أي 25% ). وبشكل أخر ففي كل مرة تحمل فيها الزوجة لديها 25% أن يولد لها
طفل مصاب و 75% أطفال غير مصابون ولكن بعضهم قد يكون حاملا للمرض(النسبة
تكون 2:3 أي 66 %) .
راجع مربع بينت للوراثة المتنحية للاب و الام حاملين للمرض
الزوج حامل للبيتا ثلاسيميا و الزوجة حاملة للانيميا المنجلية:
لو تزوج زوجين احدهما حامل للانيميا المنجلية و الاخر حامل للبيتا ثلاسيميا
فان أمام هذه العائلة أربع احتمالات في كل مره تحمل فيه الزوجة باذن
الله. الاحتمال الأول من هذه الأربعة طفل سليم من المرض(1:4 أي 25% ) و
الاحتمال الثاني طفل حامل للانيميا المنجلية (1:4 أي 25% ).و الاحتمال
الثالث هو طفل حامل للثلاسيميا (1:4 أي 25% ).و الاحتمال الرابع هو طفل
مصاب بالانيميا المنجلية و البيتا ثلاسيميا و يظهر المرض كانه انيميا
منجلية متوسطة الشدة (1:4 أي 25% ).
التشخيــــــص :
الهيموجلوبين وفقر الدم الناتج عن تكسر الدم:
الهيموجلوبين عبارة عن بروتين موجود في كريات الدم الحمراء وظيفته نقل
الأكسجين من الرئة إلى كافة خلايا الجسم ،وهو يتكون من الهيم (مادة الحديد +
صبغه) و الجلوبين (البروتين). وهناك أنواع مختلفة من بروتينات الجلوبين
المهمة منها الألفا ،البيتا ، الدلتا و الجاما جلوبين .ويتغير نوع
الهيموجلوبين في الجسم خلال الحمل إلى عدة شهور بعد الولادة نتيجة لتغير
نوع الجلوبين المنتج:
فعند الولادة يكون اغلب الهيموجلوبين(بنسبة تزيد عن 80%) من ما يعرف باسم
الهيموجلوبين الجنيني ( Hb F) ويستمر تواجده إلى أن تتم عملية إنتاج
الهيموجلوبين البالغين( Hb A) بشكل منتظم خلال الستة شهور الأولى من
العمر.,كما يوجد نسبة قليلة (1.5-3.5%) من هيموجلوبين يسمى بالهيموجلوبين
البالغين نوع 2 (( Hb A2) ).
المعدل الطبيعي للهيموجلوبين عند البالغين الذكور = 13-16 جرام/ 100 ملليليتر وعند البالغات الإناث = 12-15 جرام/ 100ملليليتر .
طريقة التشخيص
في العادة يكون لدى الشخص المصاب بالانيميا المنجلية اعراض لانخفاض
للهيموجلوبين( شحوب في البشرة)مع اصفرارا في البشرة(يرقان)و تضخم في الطحال
. و قد تنتفخ اليدين و القدمين ومع بكاء و تعب نتيجة لالم في العظام خاصة
في السنوات الاولى من العمر.و قد تكون حالة الطفل حرجة اذا كان مصحوب
بالتهاب بكتيري في الدم.ان و جود هذه الاعراض بحد ذاتها غير كافي لتشخيص
المرض و لذلك يجب اجراء ثلاثة تحاليل لتشخيص المرض:
:1-تحليل صورة الدم الكاملة( Comple Blood Count CBC):
فالشخص المصاب يكون مستوى الهيموجلوبين منخفض(بين 7 الى 10 مليغرامات لكل
100 مليلتر).و يكون حجم الكريات الحمراء و مستوى كريات الدم البيضاء و
الصفائح الدمويوة طبيعي.بينما الشخص الحامل للمرض فان هذا التحليل يكون
سليم،اي ان مستوى الهيموجلوبين غير منخفض.ولذلك هذا التحليل لا يفيد في
اكتشاف الشخص الحامل للمرض.
2- تحليل "تمنجل" الدم( sickling test) :
و هذا تحليل بسيط يتم عن طريق تعريض شريحة زجاجية عليها عينة من الدم الى
مادة مؤكسدة فظهر تحت المكبر الخلايا المنجلية بوضوح. وهذا التحليل من اهم
التحاليل التي يمكن اجرائها على عدد كبيرمن الناس لمعرفة من لدية خلايا
منجلية ام لا.وكل شخص الذي لديه خلايا منجلية بهذا التحليل قال له ان
تحليلة ايجابي المنجلي( Positive sickle test).و كل شخص لدية هذا التحليل
ايجابي يكون حامل للانيميا المنجلية او مصاب . ولذلك فهذا التحليل لا يفرق
بين الشخص الحامل للمرض و المصاب.ولذلك يجب اجراء التحليل رقم ثلاثة.
3- اختبار حركة الهيموجلوبين الكهربائية:
و هذا التحليل متوفر في الكثير من المستشفيات و هو يعطي قياس لنسبة
الهيموجلوبينات المجتلفة في الدم.و هم هيموجلوبين في حالة الشخص المصاب
بالانيميا المنجلية هو نسبة الهيموجلوبين المنجلي( HBs)فاذا كانت النسبة
اقل من 50% فالشخص حامل للانيميا المنجلية اما اذا كانت اعلى من هذه النسبة
فالشخص مصاب بالانيميا المنجلية. و اذا لم يوجد الهيموجلوبين فالشخص غير
حامل و لا مصاب بالانيميا المنجلية.
العـــــــلاج :
مرض الانيميا المنجلية من الامراض الزمنة التي لا يمكن الشفاء منها الا ان
يشاء الله.و لذلك من المهم الاهتمام بنصائح الاطباء و ارشاداتهم و
المتابعة المستمرة مع طبيب تثقون به .لقد تطور العلاج لهذا المرض في الفترة
الاخيرة فدواء الهيدروكسي يوريا و زراعة نخاع العظم و العلاج الجيني من
الامور التي لها مستقبل في مكافحة هذا المرض و التقليل من المشاكل الصحية
التي قد تصاحب هذا المرض .
من حسن الحظ ان العديد من المشاكل يمكن منعها او التقليل من شدتها.هناك
اجراءات لو اتخذت باذن الله تساعد المصابين بهذا المرض على ان يحيوا حياة
طبيعية و هنيئة .
نصائح عامة:
1- هناك أدوية معينة يجب أخذها بشكل منتظم ودقيق وعدم التوقف عن ذلك مهما
كنت الأسباب ، ومنها المضاد الحيوي ( بنسلين مثلاً ) وذلك عن طريق الفم
مرتين في اليوم حتى يصل عمر المريض إلى أكثر من 6-7 سنوات دواء حمض الفوليك
ا( Folic cid) وذلك حبة يومياً(5 مليجرام) باستمرار وذلكلزيادة الحاجة له
نتيجة لنشاط نخاع العظم لتعويض الانحلال المستمر لكرات الدم الحمراء .
2- يجب إعطاء الشخص المصاب بهذا المرض تطعيمات أخرى إضافية غير التطعيمات
المعتادة مثل تطعيم ضد جرثومة الألتهاب الرئوي نيموكوكيص( Pnumococus
vaccine ) وتطعيم ضد جرثومة هيموفيلس أنفلونزا (إHemophilus Influanza B)
وتطعيم ضد الجرثومة المسببة لالتهاب السحايا ـ الحمي الشوكية ميننجوكوكال (
meningococus) . وعند إتباع ما ذكر في الفقرتين 1و 2 فإن نسبة حماية الشخص
من الالتهابات البكتيرية القاتلة تكون عالية جداً بمشيئة الله .
3- يجب على الطبيب المعالج تدريب والدي المريض المصاب على معرفة طريقة قياس
حجم الطحال وتسجيل ذلك باستمرار وخاصة عند ملاحظة أي تغير مفاجئ في حالة
الطفل مثل شحوبه اللون ، اصفرار العينين بشكل أكبر عن المعتاد أو خمول وعدم
النشاط .
4- تناول غذاء صحي متوازن وبشكل جيد يحتوي على المجموعات الغذائية المختلفة .
5- تناول كمية من السوائل بشكل يويمي مستمر.و تزاد الكمية عند حدوث الاحمى او الاسهال او القيء او الم في العظام.
6- في فصل الشتاء ، يجب الاهتمام بتدفئة الجسم بشكل مستمر وخاصة أطراف الجسم ( اليدين والساقين ) .
7- عدم لبس ملابس ضيقة قد تضغط على الأوعية الدموية الدقيقة مما يؤدي إلى حدوث مضاعفات ( تحول الخلايا الحمراء إلى الشكل المنجلي ) .
8- الاهتمام بإعطاء الجسم قسطاً كافياً من الراحة وعدم مزاولة النشاطات بشكل كبير.
9- العناية بالأسنان بشكل مستمر وذلك بتنظيف الأسنان بشكل منتظم بعد كل
وجبة ومراجعة عيادة الأسنان بانتظام حيث أن أي تسوس في الأسنان قد يؤدي إلى
التهابات خطيرة .
10- ممارسة التمارين الرياضية بشكل معتدل وعدم الإفراط فيها .
11- تجنب تسلق الجبال العالية أو ركوب الطائرات غير مكيفة الضغط حيث أن
نسبة الأوكسجين قليلة مما قد يسبب الآلام في العظام وغيره من الجسم .
12- الحرص على المحافظة على المتابعة الدائمة للمواعيد بشكل منتظم ودقيق
وذلك في عبادة أمراض الدم وعند عدم التمكن من الحضور في الموعد المحدد يجب
الحصول على أقرب موعد ممكن والحرص على عمل التحاليل المطلوبة من الطبيب قبل
موعد العيادة .
13- إخبار المدرسة عن طبيعة مرض الطفل المصاب بفقر الدم المنجلي ولا مانع
من الاتصال بالطبيب من قبل إدارة المدرسة حتى يتمكن من تزويد المدرسة
بالمعلومات الكافية عن المرض وذلك حتى تكون المدرسة على إدراك كامل بالمرض
ومضاعفاته واكتشافها مبكراً .
14- المتابعة الطبية في المستشفى مع طبيب امراض الدم او طبيب اطفال متمرس في هذا المرض.
طرق الوقاية من حدوث نوبات انسداد للاوردة و اشعيرات الدموية(نوبات الالم):
منع الجفاف و ذلك باتناول كميات كبيرة من السؤال.
ينصح ان يتم و ضع علبة ماء (لتر و نصف )للاطفال اللذين يتجاوز عمرهم
الثالثة في الثلاجة و على الاهل متابعة و تشجيع الطفل على ان تناول هذه
الكمية خلال 24 ساعة.يمكن الاستعاضة عن الماء باي مادة سائلة كالعصير او
المشروبات الاخرى.عند حدوث اارتفاع في درجة الحرارة(حمى)يجب زيادة هذه
الكمية الى تقريبا 2 لتر او 2 و نصف ان امكن.عن حدوث اسهال فيجب اعطاء كوب
ماء (250 ميليلتر)عن كل مرة يسهل فيها الطفل.و يفضل ان يستبدل الماء في
حالة الاسهال بمحلول الجفاف المتوفر في الصيدليات او تحضيرة في المنزل اذا
تعذر الامر.يوجد في الصيدليات محاليل جفاف بطعم العصير يمكن استعمالة اذا
حدث صعوبة في اقناع الطفل على تناول السوائل.اذا حدث ارتفاع في درجة
الحرارة او اسهال و رفض الطفل تناول السوائل عليك بالتوجة الى
المستشفىلاعطاء شوائل عن طريق الوريد.
ان الجفاف و نقص السوائل تزيد من التمنجل في الدم والتي بالتالي تؤدي الى انسداد العروق ثم يتبعها الام مبرحة في العظام.
عليك الاحتياط بتناول الكثير من السوائل في الحالات التالية:
1-التعرق الشديد مع الجفاف (كالحمى)
2- الإجهاد و العب
3- الإلتهابات بجميع أنواعها خاصة المصوبة بارتفاع في درجة الحرارة.
4- الإسهال أو الاستفراغ الشديد المؤدي إلى الجفاف .
متى يجب الذهاب الى الطبيب :
يجب احضار الطفل الى الطبيب في الحال إذا كان مصابا بأحد من الاعراض التالية:
1- ارتفاع في درجة حرارة الجسم .
2- الألام في العظام او في الصدر يزيد في الشدة او لا يزول .
3-ألم شديد في البطن او انتفاخ مفاجئ وحاد في البطن .
4- شحوب مفاجئ في لون الوجه .
5- ضيق في التنفس او الم في الصدر
6- زيادة حادة في اصفرار الجسم وخاصة في العينين .
7- تضخم شديد ومفاجئ في الطحال .
8- صداع مفاجئ شديد أو تغير في مستوى الوعي ( الانتباه ) ،او تشنج
9- الآم مفاجئة شديدة في الجهاز التناسلي الذكري .
10-أسهال أو قيء
11-ءزيادة اصفرار العينين بشكل و اضح
12- ضعف مفاجئ في الاطراف
12- زغللة في العينين او ضعف مفاجئ في النظر.
علاج حالات الالم في العظام:
ان الوقاية من هذه النوبات هي العلاج الانجع و الاسهل.ولكن لو حدثة هذه
النوبات فحرص على ان يتلقى الطفل كفايتة من الادوية المسكنة للالم اضافة
الى الكثير من السوائل:
تناول الكثير من السوائل:
كما ذكرنا سابقا يجب ان تكون الكمية كبيرة و هي حوالي ضعف الكمية الذي يتناولها الطفل في العادة من دون ان يكون مريض.
الأدوية المسكنة للالم:
1- ان دواء خافظ الحرارة ايضا يخفف الالم اذا كان خفيف .
و جرعة هذا الدواء يختلف حسب وزنا لطفل.فالاطباء يعطون 10 مليجرامات من
البراسيتامول لكل كيلوجرام من وزن الطفل.ان معظم الادوية الخافظة للحرارة
عبارة عن مادة اسمها البراسيتامول( Paracetamol) . و يسوق هذا الدواء
باسماء(ماركات) مختلفة( الأادول ، البنادول، الفيفادول،التمبرا،التيلنول،و
غيرها).وكلها تحتوي نفس المادة من البراسيتامول.كما انه يمكن اعطاء الدواء
على شكل اقراص او شراب او تحميلة شرجية.و كلها لها نفس المفعول.يعطى
الدواء كل اربع الى ست ساعات ..الجرعة حسب العمر هي تقريبا 62.5 مليغرام
لاقل من 12 شهر،125 مليغرام لسن 1-4 سنوات،و 250 مليغرام لسن 4-10 سنوات،و
500 مليغرام 10-14 سنة،و 1000 مليغرام لمن هو فوق 15سنة.
ا2- ذا استمر الالم و الطفل يتناول البراسيتمول فانه يجب اعطاء دواء اقوى.
دواء الكودين فسفيت(Codeine phosphate ).و لا يمكن اخذه الى بوصفة من
الطبيب او في المستشفى.و يعطى بشكل منتظم كل 6-8 ساعات على حسب شدة الالم.و
الجرعة هي 1 او 2 مليجرام لكل كيلو جرام من وزن الطفل. و هو يفيد للالم
المتوسط الشدة،و ينصح ان يتناول مع البراسيتامول ليزيد المفعول. اذا لم
يتوفر دواء الكودين يمكن تجربة دواء ديكلوفيناك ( Diclofenac) , و يسمى
تجاريا بالفولتارين (Voltaren).و الجرعة هي 1 مليجرام لكل كيلو جرام من و
زن الطفل. و يمكن ان يعطى 3 مرات في اليوم. و يوجد منه اقراص او تحميلات
شرجية او حقن عضلية او و ريدية.
3- اذا لم تفد الادوية اعلاها او كان الالم شديد فيجب اعطاء دواء
المورفين(morphine ) او بيثيدين ( Pethidine)عن طريق الوريد اما عن طريق
جرعات متفرقة او مستمرة .في المستشفى و يجد طريقة تسمح للطفل الكبير ان
يتحكم في الجرعة التي يتناولها عن طريق ضغاط تحكم في الممغذي الذي عن طريقة
يمر الدواء.
وسائل علاج الأنيميا المنجلية: تحت الانشاء
العلاج بعقار هيدروكسي يوريا (Hydroxyurea)
في شهر يناير من عام 1995 أوقف الباحثون المشتركون في دراسة لمعرفة هل"
الهيدروسكي يوريا مفيد لتقليل نوبات الألم للمصابين بمرض الأنيميا
المنجلية" بحثهم بعد أن أظهرت النتائج إن الهيدروكسي يويا يقلل نوبات الألم
و عدد مرات الدخول إلى المستشفى بنسبة 50 % مقارنة بمجموعة أخرى من المرضى
الذين أعطوا دواء غير فعال(Placebo) للمقارنة .لقد أوقفوا البحث و الذي
اشترك فيه 21 مركز طبي قبل أربعة اشهر من نهايته أن يكملوه و ذلك للنتائج
الغير متوقعة في نجاح الدواء في التقليل من عدد نوبات الألم و يعرف ذلك
البحث بين الاطباء ببحث الهيروكسي يوريا ذو المراكز المتعددة (Multicenter
Study of Hydroxyurea (MSH Trial)).
اجري ذلك البحث على البالغين و الذين أعمارهم فوق الثامنة عشرة. و قد
وافقة الوكالة الأمريكية للغذاء و الدواء (FDA)على التصريح باستخدام هذا
الدواء لعلاج الأنيميا المنجلية.
أما بنسبة للأطفال فقد اجري بحث على 50 طفل لمن أعمارهم بين 5 إلى 15 سنة و
قد اثبت هذا الدواء فائدة في تقليل عدد مرات نوبات الألم كما هو الحال في
الكبار مع عدم ظهور أعراض جانبية لهذا الدواء على النمو أو على أعضاء الجسم
خلال مرور سنة من تناول هذا الدواء.و لكن الوكالة الأمريكية للغذاء و
الدواء لم تصرح إلى الآن باستعمال هذا الدواء للأطفال حتى تنتهي بقية
الأبحاث المتعلقة بالآثار الجانية لهذا الدواء. و قد وصلت هذه الأبحاث إلى
المستوى الثالث و يتوقع أن تظهر النتائج في خلال السنوات القادمة.
كما اجري بحث أخر على الأطفال الذين تتراوح أعمارهم بين الستة اشهر و 24
شهر و أظهرت تلك الدراسة أن هؤلاء الأطفال يتقبلون الدواء بشكل جيد.
و يعتقد الكثير من الأطباء أن هذا الدواء قد يمنع حدوث تلف في أعضاء الجسم
المتفرقة كالقلب و الكبد و و الغدد الصماء و الجهاز العصبي و لذلك فهم
يحاولون أن يقوموا بالمزيد من الأبحاث لإثبات هذه الاعتقادات.
كان و مازال دواء الهيدروكسي يوريا يستعمل في علاج أمراض الدم المرتبطة
بنشاط فمفرط و غير فعال في نخاع العظم كمرض البولي سيثيميا (polycythemia
vera) و الأورام التي لها علاقة بالدم و العقد اللمفاوية.و هذا الدواء لا
يستخدم للشفاء من الأنيميا المنجلية بل هو فقط يساعد في تقليل نوبات الألم و
التي تمثل المشكلة الكبر للمصابين بهذا المرض. لا يعرف الأطباء بشكل دقيق
كيف يعمل هذا الدواء في تقليل تلك النوبات و أن كان بعض العلماء يعتقدون أن
ذلك يحدث عن طريق رفع مستوى هيموجلوبين الجنين ( Hb F fetal hemoglobin) و
هذا بالتالي يقلل من تلاصق كريات الدم الحمراء المنجلية مع بعضها البعض و
لا يحدث انسداد في الشعيرات الدموية.كما انه لوحظ أن هذا الدواء يرفع هرمون
الاريثروبيوتين و الذي تفرزه الكلية لتنشيط و حث نخاع العظم لتصنيع
المزيد من كريات الدم الحمراء.كما إن هذا الهرمون أيضا يرفع مستوى
الهيموجلوبين الجنين ، و هذا ما جعل الأطباء يحاولون استخدام هذا
الهرمون(عن طريق إعطائه عن طريق الوريد) مع الهيدروكسي يوريا لعلاج هذا
المرض.
قد لا يفيد هذا الدواء جميع المصابين بالأنيميا المنجلية حيث أن هذا
الدواء استعمل لمن يعانون من نوبات الم متكررة و شديدة.و لذلك قد لا يستعمل
لمن حالتهم اقل شدة و ذلك لان هذا الدواء يصنف على انه من الأدوية التي
تستخدم للأورام و السرطان ( Cytotoxic Drug ) . كما أن الفائدة من الدواء
لا تظهر إلا بعد استعماله لفترة من الزمن لا تقل عن شهر بأية حال و على
المريض أن يستمر في تناوله إلى أن يقرر الطبيب غير ذلك.و
و يعطى الهيدروكسي يوريا عن طريق الفم و يبدأ بجرعة قليلة ثم تزاد الجرعة
تدريجيا إلى أن تظهر الفائدة او تظهر الأعراض الجانبية و التي تتمثل في
انخفاض كريات الدم البيضاء او الصفائح،و في هذا الحال يوقف الدواء مؤقتا ثم
يعاد البدء في إعطائه مرة أخرى بجرعة اقل.
العلاج بهرمون الاريثروبيوتين( erythropoietin )
العلاج باالجيني (Gene Therapy)
نقل دم
ينقل الدم عن الزوم و عندما ينخفض مستوى الهيموجلوبين عن 7 مليغرام لكل لتر
. و قد يستدعي نقل الدم لبعض الحالات خاصة قبل اجراء العمليات الجراحية او
عند حدوث انسداد في العروق الدموية في الرئتين . كما يقوم الطبيب بنقل
الدم بشكل دوري و مستمر كل 3-4 اسابيع لك الاشخاص الذين سبق ان اصيبوا بشلل
نصفي ناتجة عن جلطة في المخ.
العلاج بالديسفسرال
يترسب الحديد في جسم المريض تدريجيا مما يؤدى إلى تعطيل وظائف الخلايا
وبالتالي إلى موتها وفقدان وظائفها، كما يؤدى إلى الكثير من اختلال الوظائف
الهرمونية و القلبية و الكبدية و الجلدية. لذا يجب التخلص من الحديد
الزائد (اكثر من 1000 مج\مل). الديسفرال عبارة عن مادة ترتبط مع الحديد في
الجسم وتخرج مرتبطة بالحديد لخارج الجسم عن طريق البول، ومن هنا تأتى أهميه
الديسفرال الذي يحقن عن طريق الجلد لفتره8-10 ساعات يوميا او عن طريق
الوريد،والعضل
إزالة الطحال
للطحال دور هام في التخلص من كريات الدم الميتة نتيجة مشاركة الطحال في
تعويض الجسم عن عجز نخاع العظم واستمرار تراكم الحديد بصوره كبيره يبدأ
بعدها الطحال بتدمير كريات الدم الحمراء والبيضاء والصفائح الدموية ،تتفاقم
حدة فقر الدم وتتناقص مناعة ومقاومة المريض ويصبح عرضة للنزف، ويتم
استئصال الطحال عندما تظهر المؤشرات التالية:
عندما يحتاج المريض إلى نقل كميات من الدم تزيد بمقدار واحد ونصف عن الكميه الكافية.
نقص حاد في كريات الدم البيضاء أو الصفائح الدموية نتيجة تدميرها في الطحال
التضخم الكبير والمتزايد للطحال.
زرع نخاع العظم (Bone Marrow Transplant)
هذه العملية تعتمد على وجود متبرع يفضل أن يكون من أشقاء او شقيقات المريض
حيث يجب أجراء الفحوصات التي تؤكد من وجود التطابق النسيجي والخلوي (100%)
بين المتبرع والمريض،وتزداد فرص نجاح العملية للذين لا يعانون من ترسب
الحديد أو تضخم الكبد والطحال.
إن رفض الانسجه عادة ما يكون اقل نسبة في الأطفال عنه في الكبار فذلك لا
يحدث مباشرة بعد العملية إنما قد يحتاج إلى 3-5 سنوات يبقى فيها خطر
الرفض،فالمريض بحاجة إلى متابعة دقيقه للغاية.
ويتم فبل زرع نخاع العظام التخلص من نخاع المريض بتناوله الأدوية تدمر
خلايا النخاع وعلى مدى 6 أيام يشعر فيها بالوهن والضعف وتحت التعقيم التام
يتم شفط النخاع بواسطة حقنه خاصة وتحت التخدير العام وتستغرق هذه العملية
حوالي نصف الساعة وتستكمل العملية بإعادة حقن نخاع العظام السليم إلى
المريض فيما يشبه عملية نقل الدم فيجد النخاع الجديد تجويف نخاع المصاب.
والمتبرع عادة لا يعاني من أي مخاطر وجسمه قادر على تعويض الكميه التي تم
شفطها من نخاع العظام ولا يحتاج للبقاء في المستشفى اكثر من يوم واحد فقط.
الجديد في علاج الانيميا المنجلية:
بدأت بعض المراكز في إجراء عمليات نقل وزراعة نخاع العظم للجنين المصاب
داخل رحم أمه بدل إجهاضه وتتميز هذه العملية بندرة رفض جسم المريض للنخاع
المزروع.
طرق الوقاية:
فحوصات قبل الولادة
إذا كان كلا الوالدان يحملان الانيميا المنجلية فمن الممكن أن يتم فحص
الجنين للتأكد من سلامته من الاصابة بالانيميا المنجلية خلال الأسبوع
10-11 من الحمل. ويتم الفحص بأخذ عينه من المشيمة ( Chorionic Villus
)وتحليلاها بواسطة ال DNA لمعرفة إذا ما كان الجنين مصاب أم لا.،وتظهر
نتيجة التحليل خلال 3-4 أسابيع.وكما هو الحل في جميع الفحوصات الطبية فقد
يكون هناك بعض المضاعفات واهما احتمال الإجهاض من جراء سحب العينه 1%لثانية
هي عن طريق سحب من السائل الأمينوسي والموجود حول الجنين.وتأخذ هذه العينة
في الأسبوع 14 -16 من الحمل واحتمال الإجهاض في هذه الحالة حوالي نصف في
المائة 0.5%
فحوصات قبل الزواج :
يمكن الكشف على أي شخص لمعرفة إذا ما كان حامل للمرض و بطريقة سهلة و ميسرة
و متوفرة في معظم المختبرات الطبية .و هو تحليل رخيص في قيمته المادة لكنه
غالي في قيمته العلمية. ولا يتوقع أن يكلف هذا التحليل أكثر من 15
دولار.و ينصح بإجراء هذا التحليل لكل من له أصول عرقية بمنطقة الخليج
العربي و جنوب السعودية و اليمن و المناطق الشمالية الغربية من السعودية. و
إن كنا نفضل أن يجرى هذا التحليل إضافة إلى تحاليل المتعلقة بالثلاسيميا
على جميع من يريد الزواج بغض النظر عن أصولة العرقية و ذلك لرخص قيمته و
توفره .
DREAM MAN- مدير موقع المعهد الطبي
- تاريخ التسجيل : 25/01/2010
عدد المساهمات : 533
المزاج : رايق
الجنس :
رد: امراض الدم الوراثية بالتفصيل و بالعربي
من طرف DREAM MAN الخميس يناير 27, 2011 11:48 pm
انيميــــــــــــا الفــــول :
يعتبر مرض انيميا الفول اكثر امراض الأنزيمات انتشارا في العالم.فهو يصيب
حوالي 400 مليون شخص ولو نظرنا الى التوزيع الجغرافي للمرض لوجدنا انه
ينتشر في مناطق كانت موبؤه بمرض الملاريا.و مرض الملاريا من الأمراض
الفتاكة و التي يسببها طفيل اسمة طفيل الملاريا.ويعيش طفيل الملاريا متطفلا
على كريات الدم الحمراء فهو يستخدمها في احد اطوار حياته،و في كثير من
الأحيان يؤدي الى تكسيرها وتحللها .ويبدوا ان جسم الأنسان "تأقلم" مع هذا
المرض عن طريق جعل الكريات الحمراء تقاوم استيطان طفيل الملاريا فيها و ذلك
بإحداث طفرة في جين انزيم G6PD فيجعل كريه الدم الحمراء تتكسر و تتحلل
عند تعرضها لالتهاب بطفيل الملاريا، و بذلك يلا يستطيع الطفيل اكمال دورة
حياته التي يستلزم العيش داخل كريه الدم الحمراء لبعض الوقت،و بذلك يتخلص
الجسم من الملاريا بشكل فعال.وبعد ان اختفى المرض من كثير من مناطق العالم
بقية الطفرة على حالها ولم يرجع الجين الى حالته السابقة.وبما ان مرض
الأنيميا المنجلية و مرض الثلاسيميا ايضا يعتقد انها امراض تنتشر في
المناطق الموبؤه بمرض الملاريا فإنه ليس من الغريب ان يصاب الشخص بهذه
الأمراض.فبعض المصابون بمرض الأنيميا المنجلية او الثلاسيميا ايضا مصابون
بمرض انيميا الفول.
يعرف المرض بين الأطباء بمرض نقص خميرة (انزيم) ديهيدروجينيز الجلوكوز 6
فوسفاتي(GLOCUSE 6 PHOSPHATE DEHYDROGENASE )او بالمختصر ( G6PD). يعبتر
هذا المرض مرض وراثي نتيجة لطفرة موجودة على كروموسوم اكس فلذلك يعتبر من
الأمراض الوراثية التي تنتقل بالوراثة المرتبطة باالجنس.وهو في العادة يصيب
الذكور و ينتقل من امهاتهم .وفي بعض الأحيان قد يظهر المرض على الاناث كما
ان الذكور المصابون بالمرض ينقلون المرض ولكنهم ينقلونه الى بناتهم ولا
ينقلنهوا الا ابنائهم مطلقا.ونقص اللأنزيم يجعل الكريات الدم الحمراء معرضه
للتحلل والتكسر قبل موعدها المعتاد(والذي في العادة يتجاوز 100 يوم) فيؤدي
الى انخفاض في الهيموجلوبين (فقر دم او انيميا)مع انتشار للمادة الصفراء
تعجز عن تصفيتة الكبد بشكل سريع.هناك تفاوت كبير في السن الذي تظهر فيه
اعراض المرض.فقد يظهر عند المواليد على مباشرة بعد الولادة فيكون اليرقان
عندهم اعلى من المستوى المعتاد و الذي يصيب الكثير من الأطفال الطبيعين كما
انه قد يحدث في أي سن و لكنه في العادة يظهر عند ما يتناول المصاب بالمرض
الفول او العدس او أي نوع من البقوليات او بعد الأصابة بمرض فيروسي او عند
تناول بعض من العقاقير.كما قد تظهر الاعراض من دون ان يصاب الشخص بأي مرض و
من دون ان يناول أي نوع من المواد المؤكسدة كلبقوليات.
الاعــــراض :
ليس هناك اعراض خاصة بمرض انيميا الفول.ولذلك تشترك اعراض هذا المرض
بالعديد من الأمراض التي تسبب تكسر او تحلل في في كريات الدم اللحمراء.
وهذه الاعراض هما فقر الدم(انيميا) و اليرقان(اصفرار الجلد و االعينين )وقد
يحدث تكسر في كريات الدم الحمراء بشكل حاد (مفاجئ) او بشكل مزمن
(ببطء).ولذلك يمكن ان نقسم اعراض المرض الى هذين القسمين
التكسر الحاد :
يسبب تكسر الكريات الحمراء الى انخفاض نسبة الهيموجلوبين في الدم.ولذلك
فالتعريف العلمي لفقر الدم او الأنيميا هو انخفاض مستوى الهيموجلوبين الى
مستوى اقل من المعدل الطبيعي (المعدل الطبيعي يختلف حسب العمر و الجنس
ولكن هو دائما فوق 10 غرامات لكل 100 مليمتر من الدم).و يكون لون الشخص
المصاب بفقر الدم شاحب و يستدل الطبيب الى ذلك بفحص الغشاء المبطن للجفن
السفلي للعين.فكلما قل الون الأحمر في الجفن كلما دل على انخفاض في مستوى
الهيموجلوبين .و عند تكسر كريات الدم الحمراء تنتنج مادة صفراء تعرف
بالبيلوروبين ( Bilirubin) وتيتخلص منها الجسم عن طريق الكبد.ولكن اذا
ارتفع مستوى البيلوربين في الدم(نتسجة لتكسر عدد كبير من كريات الدم
الحمراء دفعة واحدة) فانه يرشح والى الجلد وبقيت الاعضاء ويظهر لون الجلد و
العينين اصفر.و ارتفاع مادة البيلوربين في العادة ليست مضرها الا في
المواليد نظرا لضعف الجدار العازل لشرايين في المخ في ذلك العمر.ولذلك يجب
اخذ الأمر بمحمل من الحرص و الأهتمام في حالة اصفرار جلد حديثي الولادة.
في بعض انواع نقص انزيم G6PD يكون تكسر او تحلل كريات الدم الحمراء بطيء و
لا تتكسر الكريات بشكل مفاجىء.هذا النوع من التكسر يسبب فقر دم مزمن وقد
يكون مصحوب باصفرار بسيط في الجلد.و قد لا يحدث فقر دم اذا استطاع الجسم
انتاج كمية اعلى من الهيموجلوبين تعوض عن النقص المستمر.يحتاج الشخص المصاب
بهذا النوع من التكسر تغذية جيدة خاصة من فيتامين حمض الفوليك والذي يمكن
تناولة على شكل اقراص او شراب اذا لزم الامر.
في بعض الأحيان يحدث التكسر لأسباب مجهولة ولكن بشكل عام ان تعرض الجسم لأي
مادة مؤكسدة يمكن ان تكسر الدم. واليك بعض من اهم المواد المكسرة للدم
والتي ينصح بتجنبها :
1- تناول بعض الاطعمة:وهذي الأطعمة هي البقوليات بجميع انواعها خاصة
الفول و العدس و البازلاء والفاصوليا.و تتراوح كمية المادة المؤكسدة بين
نوع و اخر من الأطعمة. وقد كون الكمية التي يتناولها الشخص قليلة فلا تسبب
له مشكلة ولكن في الكثير من الأحيان يتناول الشخص كمية قليلة فتسبب له تكسر
حاد في الدم وا حياننا يتناول نوع معين من الأطعمة لسنوات عديدة ولا تسبب
له تكسر وفجاءة تتكسر لديه الكريات بعد تناول كمية قليلة منه.و نظرا للحاجة
لنقل الدم لبعض الأشخاص الذين انخفض لديهم مستوى الهيموجلوبين بشكل حاد
،فإننا ننصح جميع المصابين بمرض انيميا الفول تجنب جميع الاطعمة المتعارف
انها تسبب تكسر في الدم وعدم التهاون في هذا الأمر نظرا للمخاطر التي قد
يتعرض لها الشخص عند نقل الدم له خاصة من الأمراض المجهولة .
2- تناول بعض انواع من الأدوية:على كل المصابين بهذا المرض تنبية الطبيب
المعالج على انهم مصابون بهذا المرض لكي يتفادة اعطائهم بعض انواع من
الأدوية و استبدالها بانواع مأمونه،الا ذا كان الشخص مصاب بالملاريا و يضطر
الطبيب على استعمال دواء محدد لهذا المرض.(يمكن الاطلاع على هذه الأدويه
في صفحة العلاج).
3- التعرض للالتهابات الفيروسية او البكتيرية.
الاسبـــــــــاب :
ينتج انزيم G6PD جين موجود على كروموسوم أكس يعرف بجين انزيم G6PD.و حين
يتعطل هذا الجين يحدث مرض انيميا الفول.ولذلك يعتبر هذا المرض مرضاً من
امراض الجينات الوراثية.
يحدث في داخل الخلية الحمراء عدة تفاعلات كيميائية و على شكل شبكة مترابطة
من المواد الكيميائية تتفاعل مع بعضها البعض و مدعومة بالعديد من
الأنزيمات.و لو ركزنا على احد هذه الشبكات و المسمية التفاعل فسفات
البنتوز.ودور انزيم G6PD هو انتاج مادة البنتوز من جلوكوز الفسفات السداسي و
تحويل مادة النيكوتونوميد ادنين للفسفات الثنائي النووي (NADP ) الى مادة ا
لنيكوتونوميد ادنين للفسفات الثنائي النووي المؤكسد( NADPH).يوجد عدة طرق
اخرى لأنتاج مادة ( NADPH) في جميع خلايا الجسم ماعدى كريات الدم
الحمراء،فتفاعل فسفات البنتوز هي الطريقة الوحيد لأنتاج ( NADPH). يكمن
اهمية مادة ( NADPH)في انه يحافظ على جعل مادة الجلتثيون في داخل كرية الدم
الحمراء في حالة مختزلة قابلة لسحب الهيدروجين من أي مادة مؤكسدة كلفول
والبقوليات و بعض العقاقير والألتهابات وحماية الكرية الدم الحمراء من
التكسر.
وظيفة انزيم G6PD:
عند نقص انزيم G6PD يؤدي لنقص تكون مادة ( NADPH) ويجعل مادة الجلتثيون في
حالة مؤكسدة(حالة الأكسدة ضد الاختزال).الا هذا الحد لا تتأثر الكريات الدم
الحمراء بهذا النقص،ولكن عندما يأكل الشخص مادة الفول فان الهيموجلوبين في
داخل الخلية الحمراء يتبلور نتيجة لعدم حمايتة من قبل مادة الجلتثيون و
بذلك تكسر الكريات الدم الحمراء مؤديتا الى فقر دم وصفار.
الوراثة و انزيم G6PD :
لا شك ان مرض نقص انزيم G6PD يعتبر مرضاً وراثية ولكنه في العادة يظهر على
الذكور ولا يظهر على الاناث .و لذي ينقل المرض الى الذكور هن الأناث و
الذكور المصابة بالمرض تنقلة فقط لاناث.قد يبدوا ان الموضوع محير او معقد
ولكن يمكن فك كل الطلاسم بمعرفة الجانب الوراثي من القصة.ولكي يسهل عليك
فهم الموضوع عليك ان تعرف بعض اساسيات الوراثة .فمن المهم ان تعرف ما معنى
الخلية والنواة و الكروموسوم والجين. ولذلك عليك الذهاب الى هذا الرابط
للتعرف على كل ذلك ثم الرجوع لتكملة قراءة الموضوع.
يحدث مرض نقص انزيم G6PD بسبب وجود طفرة على الجين المنتج لذلك الانزيم
والموجود على الذراع الطويلة كروموسوم أكس.هناك انواع كثيرة من الطفرات
المؤدية لنقص او ضعف انزيم G6PD .ويعتقد انه يوجد فوق 400 نوع من
الطفرات.واختلاف انواع الطفرات يفسر الى حد ما الاختلاف في الأعراض،و هناك
بعض الطفرات التي تتميز بإحداث تكسر تلقائي لكريات الدم الحمراء للمواليد
وهناك انواع اخرى تتميز بحدوث تكسر تلقائي مستمر للدم حتى من دون التعرض
للمواد المؤكسدة كالفول..
يقع انزيم G6PD في الشريط رقم 28 من الذراع الطويلة من كروموسوم أكس.وبما
ان المرأة لديها نسختان من كروموسوم أكس فأنها لديها أيضا نسختان من جين
إنزيم G6PD نسخة أتتها من أمها و الأخرى أتتها من أبوها.وبما أن الرجل ليس
لدية الا نسخة واحدة من كروموسوم أكس، فأنه لا يحمل الا نسخة وحيدة من جين
إنزيم G6PD.و بما أن الجينات قابلة للعطب فأن حدوث عطب في احد نسختي جين
إنزيم G6PD في المرأة لا يؤدي في العادة للإصابة بالمرض،لان المرأة لديها
نسخة إضافية على كروموسوم أكس الأخر.ولكن الرجل على خطر،فأن إصابة نسختة
الوحيدة بعطب يسبب له المرض.
قسم الأطباء الأمراض الوراثية الا أربعة أقسام رئيسية .القسم الأول الأمراض
الوراثية المتعلقة بالكروموسومات،القسم الثاني الأمراض المتعلقة
بالجينات(ويسمى أيضا بأمراض وحيد المورثة)والنوع الثالث الأمراض الوراثية
المتعددة الأسباب ،والنوع الرابع الأمراض المتعلقة بالميتوكندريا.
و يتفرع من القسم الثاني أربع أنواع من الأنماط (الطرق) الوراثية التي
ينتقل فيها المرض من شخص إلى أخر.النمط الأول يسمى الوراثة المتنحية
،الثاني الوراثة السائدة،الثالثة الوراثة المتعلقة بالجنس
المتنحية،والرابعة الوراثة المرتبطة بالجنس السائدة.يعتبر مرض انزيم G6PD
من الأمراض المتعلقة بالجينات و ينتقل بالوراثة المتعلقة بالجنس المتنحية.و
سمي بالوراثة المتعلقة بالجنس المتنحية،لان الجين المعطوب يقع على إحدى
الكروموسومات المحددة للجنس ،كروموسوم أكس.و سمي بالمتنحى لأن المرض لا
يظهر على الشخص الا اذا أصيبت كل نسخ كروموسوم أكس بالعطب.ولذلك في العادة
لا يظهر على المرأة لآن لديها نسختان من كروموسوم أكس،ولكنه يظهر على الرجل
لآنه لا يحمل الا نسخة واحدة.ولمعرفة المزيد عن الوراثة المتعلقة بالجنس
المتنحية يمكنك الرجوع لهذه الصفحة:الوراثة المرتبطة بالجنس المتنحية.
متى يصاب كلا من الرجل و المرأة للمرض و متى تصبح المرأة حاملة للمرض ؟؟
يصاب الرجل بالمرض اذا كانت النسخة التي لدية من جين انزيم G6PD معطوب(به
طفرة).و بما ان جين انزيم انزيم G6PD موجود على كروموسوم أكس و الرجل ليس
لدية الا نسخة واحدة جاءته من امة فان المرض ينقل من الام لأبنائها . و
ابنائها المصابون لا ينقلون المرض لأبنائهم لانهم لا يعطون ابنائهم كرموسوم
أكس فهم يعطونهم كروموسوم واي .فلذلك لا ينتقل المرض من الرجل للأبنائه
الذكور اطلاقاً.ولكن الرجل من المصاب من الممكن ان ينقل المرض لبناته لانه
يعطيهن نسختة المعطوبة من كروموسوم أكس الورحيد عنده.(لتعرف على الفرق بين
الانثى والذكرمن ناحية التركيبة الوراثية ارجع لهذا الرابط: تحديد الجنس).
تكون المرأة حاملة للمرض اذا كانت فقط احدى نسختي الجين الموجودة على كروموسوم أكس مصابة بالعطب.
متى تحمل المرأة المرض
تكون المرأة حاملة للمرض اذا كانت فقط احدى نسختي الجين الموجودة على كروموسوم أكس مصابة بالعطب.
تصاب المرأة بالمرض اذا اصيبت كلتا النسختين التي لديها بالعطب.فلذلك نجد
ان النسختان التي على كروموسوم أكس من الاب ومن الام معطوبة.و هذا يعني ان
ابوها ايضا مصاب بالمرض و انه على اقل تقدير ان امها حاملة او مصابة
بالمرض.اذا فهمنا هذا الأمر كقاعدة فعلينا ان نعلم ان هناك حالات استثنائية
تصاب فيها المرأة بالمرض بدون ان تكون الأم حاملة او مصابة بالمرض وقد
يحدث احياناً ان الأم فقط حاملة للمرض او مصابة بينما الأب سليم.وفي ما يلي
الحالات التي تصاب بها المرأة بالمرض:
1-عندما يكون الأب مصاب والام حاملة او مصابة بالمرض.وتكثر هذه الحالة في
المجتمعات التي يكثر فيها زواج الأقارب ،خاصة الزواج بين ابناء الخالات او
الأخوال.
2- عندما يكون الأب فقط مصاب بالمرض بينما الأم سليمة(أي ليست حاملة و لا مصابة بالمرض).
واحدة منهما عليه نسخة من جين انزيم G6PD .و لذلك فالمرأة اذا عطبت احدى
نسختيها فانها في العادة لا تصاب المرض لان لديها نسخة اخرى.ولكن هناك امر
مهم يتحتم علينا معرفته من ناحية الفروقات الوراثية بين الرجل و المرأة.كما
نعلم ان الأنسان لدية 46 كروموسوم وهي تأتي على شكل ازواج،وكل نسخة من هذه
الأزواج تأتي من الأم والأب.ولو نظرنا الى الزوجد الجنسي لوجدنا ان المرأة
لديها نسختان من كروموسوم أكس بينما الرجل لديه فقط نسخة من كروموسوم أكس
واخرى من كروموسوم واي.و مع ان المرأة لديها نسختين من كروموسوم أكس في كل
خلية الا ان كل خلية لا يعمل فيها الا نسخة واحدة فقط و تقوم الخلية بتعطيل
النسخة الثانية و ذلك لعدم حاجتها
لها،و هذا شىء متوقع لان جميع خلايا الرجل تعمل بشكل طبيعي بنسخة واحدة
لذلك فالله عز وجل جعل الخلية الأنثوية تعطل الكوموسوم الزائد لديها ولذلك
لكي لا ينتج كميه مضاعفة من البروتينات.تسمى خاصية تعطيل كروموسوم أكس في
الخلية الأنثوية بتثبيط او تعطيل أكس(X inactivation ).و بما ان كل خلية
فيها كوموسوم أكس من الأب و اخر من الأم فان الخلية لا تفرق بينهما فهي
تقوم بتعطيل الكروموسوم بغض النظر عن مصدرة.هذه الخاصية تسمى تعطيل أكس
العشوائي( Random X in activation).ولذلك لو نظرنا الا جميع خلايا المرأة
لوجدنا ان بعض خلاياها يعمل بها كروموسوم أكس من الأب والبعض يعمل بها
كروموسوم من الأم.
و بإمكان الطبيب او اخصائي المختبر ان يشاهد كروموسوم أكس المعطل ملتصق
بجدار الخلية من الداخل ويظهر في خلايا الدم البيضاء على شكل عصيه صغيرة
معروفة بعصى الطبل( Drum stick)وفي الخلايا المبطنة للفم بأجسام بار ( Bar
Bodies).وبما ان التثبيط يتم بشكل عشوائي في جميع الخلايا،فاننا نجد ان
حوالي 50% من الخلايا في جسم المرأة يعمل بها كوموسوم أكس مصدرة الأب و 50%
يعمل بها كروموسوم أكس مصدرة الأم.طبعا لا يفرق عند الخلية اذا ماكان
الكروموسوم النشط او المعطل هو كروموسوم كان مصدرة الأم او الأب لان
الجينات الموجودة على كروموسوم أكس الأنثوي او الذكري متطابقة و لا حتى
يمكن معرفة الفرق بينهما من الناحية التكوينية.ولكن الأمر مهم اذا كان احدى
هذه الكروموسومات يحمل جيناً غير سليم او به طفرة.و أليك التفاصيل.
لو قلنا ان امرأة ما اباها مصاب بمرض انيميا الفول(أي ان كروموسوم أكس الذي
لدية به جين به طفرة سببت المرض)، وان امها سليمة(أي ان كلا نسختي
كروموسوم أكس التي لديها سليمة من العطب).و على الأب و الأم ان يعطيا نسخة
من كروموسوم أكس(و هذا شرط ليكون لطفل انثى لا ذكر)وبما ان الأم لديها
نسختان من أكس وكلتاهما سليمتان فان النسخة التي سوف تعطيها لابنتها
سليمة،بينما الأب فليس لدية الا نسخة و حيدة من كروموسوم أكس وعليها جين(
لإنزيم G6PD) معطوب فلذلك ليس لديه أي اختيار الا اعطائها اياه.طبعا المرأة
تعطي كروموسوماتها على شكل بويضة بها 23 كروموسوم ،و الرجل ايضاً يعطي
كروموسوماته على شكل حيوان منوي به 23 كروموسوم.وعند اتحاد البويضة
والحيوان المنوي تكتمل عدد الكروموسومات فتصبح 46 كروموسوم وهذه الخلية
تنقسم الى بلايين الخلايا لتكون انسان كامل.لو نظرنا الا خلايا هذه البنت
لوجدنا ان جميع الخلايا بها كروموسوم أكس عليه جين انزيم G6PD سليم(اتى من
الأم) و اخر عليه جين انزيم G6PD معطوب،ولذلك تكون هذه البنت حاملة للمرض
وليست مصابة وذلك نظراً لان الجين السليم ينتج كمية كافية من انزيم G6PD
ويغطي النقص الذي احدثة الجين المعطوب.الا هنا و الأمر طبيعي وهذا الذي
يحدث في كثير من الحالات. لو نظرنا الا جميع خلايا هذه البنت لوجدنا ان 50%
من خلاياها يعمل بها كروموسوم أكس السليم(الذي اتاها من امها)بينما البقية
يعمل بها كروموسوم أكس غير السليم(الذي اتاها من ابوها).هذا هو المعتادـ.و
لكن بما ان تعطيل (تثبيط)كروموسوم أكس يتم بشكل عشوائي ،أي ان كل خلية ليس
لها علاقة بالخلية الاخرى و اذا تعطل كروموسوم أكس من الأم في خلية ما
فليس من الضرورة ان الخلية الاخرى ان يتعطل كروموسوم أكس من الأب ،فأنه ليس
من الضرورة ان تكون نسبة كروموسومات أكس من الأم ومن الأب 50% لكل
منهما.بل من الممكن ان تكون مثلا 70% من الخلايا يعمل بها كروموسوم أكس
الذي من الأم، وقد يكون اكثر من ذلك او اقل.
لا يهمنا هنا نسبة الخلايا التي يعمل بها كروموسوم أكس الأتي من الأم(أكس
السليم)بقدر معرفة نسبة الخلايا التي يعمل بها كروموسوم أكس الأتي من
الأب(أكس الغير سليم)فلكما زادة نسبة الخلايا التي يعمل بها كروموسوم أكس
الأتي من الأب لكما زاد عدد الخلايا التي لا يعمل بها جين انزيم G6PD
المعطوب.وكلما زادة عدد الخلايا في الجسم والتي يعمل بها كروموسوم أكس
المعطوب كلما زاد احتمال ان تظهر اعراض مرض نقص انزيم G6PD على هذه البنت
حتى وان لم يكن جميع خلاياها تعاني من هذا النقص، والمهم هوا النسبة بين
عدد الخلايا التي يعمل بها جين انزيم G6PD و بين الخلايا التي لا يعمل
بها.وكلما زادة النسبة لصالح الجين الذي لا يعمل كلما زادة نسبة احتمال ان
تصاب البنت بالمرض ويظهر عليها اعراضة.ان هذا الشرح المطول هو شرح لما
يعرف بظاهرة ليون(Lionization ) او تعطيل كروموسوم أكس الغير عشوائي( Non
Random X inactivation).والشرح السابق ينتبق ايضا على الحالات التي يكون
فيها الأب سليم(كروموسوم أكس الذي يحملة عليه جين انزيم G6PD سليم)والأم
حاملة او مصابة بالمرض.ففي كل مره تحمل فيها الأم بانثى فان زوجها سوف يعطي
دائما بنسخة سليمة من كروموسوم أكس.بينما الأم من اللمكن ان تعطي نسخة
سليمة او معطوبة(هذا اذا كانت حاملة للمرض)او دائما تعطي بنسخة معطوبة(لو
كانت مصابة بالمرض وكلتا نسختيها من كروموسوم أكس معطوبتان).
3- عندما تكون البنت مصابة بمرض أخر يسمى بمتلازمة تيرنر.فهذا المرض يصيب
البنات فقط وهو ناتج عن نقص في عدد الكروموسومات .فبدل ان يكون مجموع
الكروموسومات 46 يكون لديها فقط 45 كروموسوم .والناقص هو احد نسختي
كروموسوم أكس.فإذا حدث وكانت النسخة التي لديها فيه اجين انزيم G6PD معطوب
فأنها تصاب بالمرض وتشابه حالتها حالة الذكر المصاب بالمرض فهو لا يملك الا
نسخة واحدة من كروموسوم اكس وبه نسخة معطوبة من جين انزيم G6PD.
احتمالات الاصابة :
تعتمد احتمالات تكرر المرض في العائلة الواحدة حسب الحالة المرضية
للوالدين.ونظراً الا ان نقص الانزيم لا يظهر على الأناث فاننا سوف نعتمد
على الحالة الجينية للأنزيم مباشرة.و لكن على القارئ ان يعرف ان الأطباء في
العادة يقيسون كمية الانزيم في الدم و لا يجرون تحليل للجين مباشرة.و
السبب في ذلك هو صعوبة اجراء الفحص الجيني على المرضى و لا يجرى هذا
التحليل الى في الدراسات و الأبحاث الطبية
فيما يلي بعض الحالات الجينية للوالدين واحتمالات الأصابة:
الام سليمة و الزوج مصاب /
انظراً لوجود جين انزيم G6PD على كروموسوم أكس وليس على كروموسوم واي،و
نظراً الى ان الأب يعطي ابنائة الذكور كروموسوم واي ،فإن جميع ابنائة
الذكور يكونوا سليمين من المرض.بينما جميع بناتة حاملات للمرض.طبعا المرأة
الحاملة للمرض لا تصاب بالمرض و لا تظهر عليها اعراض المرض،ولكن عليك ان
تعرف انه في بعض الأحيان تظهر اعراض المرض على المرأة الحاملة للمرض،ولذلك
عليك الرجوع الى الفقرة التي تتحدث عن الحالات التي تصاب بها المرأة في
اعلى هذه الصفحة.انظر جدول تلقيح البويضات والحيوانات المنوية للأم
السليمة والزوج المصاب.
الام حاملة للمرض و الزوج سليم /
ان احتمال اصابة الذكور في هذه الحالة 50% في كل مرة تحمل فيها تحمل فيها
الأم.كذلك 50% من الأناث يكن حاملات للمرض كأمهن وفي العادة لا يصبن
بالمرض(ولكن ارجع للحالات الأستثنائية لاصابة المرأة في اعلى الصفحة). انظر
جدول تلقيح البويضات والحيوانات المنوية للأم الحاملة للمرض والزوج سليم.
الام حاملة للمرض و الزوج مصاب /
ان احتمال اصابة الذكور في هذه الحالة لا ترتفع عن الحالة السابقة فهي 50%
في كل حمل.ولكن احتمال اصابة الاناث في هذه الحالة تصل الى 25% ( أي ان كلا
نسختي جين انزيم G6PD معطوبتان)و الباقي منهن حاملات للمرض(أي لديهن نسخة
واحدة من جين انزيم G6PD معطوب و نسخة سليمة). انظر جدول تلقيح البويضات
والحيوانات المنوية للأم الحاملة للمرض والزوج المصاب.
الام مصابة و الزوج سليم /
ان احتمال اصابة الذكور في هذه الحالة هي 100% كما ان 100% من الاناث
حاملات للمرض. انظر جدول تلقيح البويضات والحيوانات المنوية للأم المصابة
بالمرض والزوج السليم.
الام و الزوج كلاهما مصاب /
و الأم في هذه الحالة مصابة بالمرض و كلا نسختي الجين من انزيم G6PD
معطوبتين .ان احتمال اصابة الذكور والأناث في هذه الحالة 100%. انظر جدول
تلقيح البويضات والحيوانات المنوية للأم و الاب المصابين.
العـــــــــلاج :
لا يوجد للأسف علاج شافي من هذا المرض،و لا يمكن التخلص منه ومنعه من
الانتقال من جيل لاخر.المرض ليس بالمعدي ولي لمخالطي المصاب او حاملي المرض
أي خطر على الأخرين .و المرض لا ينتقل بالمعاشرة الجنسية ولكن كما هو
معروف فالصفات الوراثية و يدخل في ذلك الجينات تنتقل من اتحاد الحيوان
المنوي بالبويضة .
العلاج من هذا المرض يتمحور حول تجنب تكسر الدم عن طريق تجنب التعرض للمواد
المؤكسدة كانواع معينة من الاطعمة والادوية و الالتهابات بشكل عام
الأطعمة التي يجب تجنبها
جميع البقوليات
الأدوية التي يجب تجنبها
Vitamains الفيتامينات
Vitamin K
Ascorbic acid(Vitamin C)
Anti-Malarial Drugs مضادات الملاريا
Pamaquine
chloroquine
Hydroxychloroquine
Mepacrine (quinacrine)
pamaquine
primaquine
Pentaquine
Quinocide
Quinocide
Quinaorine (Atabrine),
Quinine (this is used sometimes in soft drinks such as tonic water)Biguanides
Pyrimidines
Quinolines
Sulphonamides
Sulphones
Sulfanamides/ Sulfones مضادات السلفا
Sulfacetamide
Sulfanilamide
Salioylazulfapyridine (Azulfidine)
Sulfapyridine
Sulfasalazine
Sulfamethoxypyrimidine
Sulfaoethamide
N2 – Adetylbulfanilamide
2-Amino-5-Sulfanilylthiazole
Sulfiboxazole (Gantrisin)
Thizolsulfone (Promizole)
Diaminodipenysulfone (DDS)
Solfoxone (or Sulphoxone; Dapsone)
Anti-Inflammatory / Analgesic Drugsالمسكنات و مضادات الروماتزم
Salicylic Acid (Aspirin)
Phenacetin
Antipyrine
Aminopyrine (Pyramidon)
Aniline
Fenamic Acid
Pyrazolone (Acetanilide)
para-aminosalicylic acid
Anti-Bacterial Drugs المضادات الحيوية
co-trimoxazole
Cephalsoporins
Chloramphenicol
Nitrofurans (Furadentin)
Nalidixic acid
Furaltadone (Altafur)
Nitrofurazone (Furadin)
Penicillins
Quinolones
Chloramphenicol
Anti-Emetic / Neuroleptic Drugs مضادات التقيؤ
Phenothiazine derivatives
Uricosuric Drugs طاردت ليوريك
Benzoic Acid derivatives
Anti-Histaminic Drugsمضادات الهستامين
Antazoline
Cardiac/Anti-Hypertensive Drugs ادوية القلب و الضفط
Procainamide
Quinidine
Hydralazine
Anti-Tuberculous Drugs مضادات السل
Isoniazide
P.A.S.
Anti-Protozoal Drugsمضادت الطفيليات
Naphthalene
Miscellaneous متفرقات
Chelating Agents
Dimercaprol
Methylene blue
Naphthalene
Doxorubicin
L-dopa
انواع محددة من الأعشاب الصينية
يعتبر مرض انيميا الفول اكثر امراض الأنزيمات انتشارا في العالم.فهو يصيب
حوالي 400 مليون شخص ولو نظرنا الى التوزيع الجغرافي للمرض لوجدنا انه
ينتشر في مناطق كانت موبؤه بمرض الملاريا.و مرض الملاريا من الأمراض
الفتاكة و التي يسببها طفيل اسمة طفيل الملاريا.ويعيش طفيل الملاريا متطفلا
على كريات الدم الحمراء فهو يستخدمها في احد اطوار حياته،و في كثير من
الأحيان يؤدي الى تكسيرها وتحللها .ويبدوا ان جسم الأنسان "تأقلم" مع هذا
المرض عن طريق جعل الكريات الحمراء تقاوم استيطان طفيل الملاريا فيها و ذلك
بإحداث طفرة في جين انزيم G6PD فيجعل كريه الدم الحمراء تتكسر و تتحلل
عند تعرضها لالتهاب بطفيل الملاريا، و بذلك يلا يستطيع الطفيل اكمال دورة
حياته التي يستلزم العيش داخل كريه الدم الحمراء لبعض الوقت،و بذلك يتخلص
الجسم من الملاريا بشكل فعال.وبعد ان اختفى المرض من كثير من مناطق العالم
بقية الطفرة على حالها ولم يرجع الجين الى حالته السابقة.وبما ان مرض
الأنيميا المنجلية و مرض الثلاسيميا ايضا يعتقد انها امراض تنتشر في
المناطق الموبؤه بمرض الملاريا فإنه ليس من الغريب ان يصاب الشخص بهذه
الأمراض.فبعض المصابون بمرض الأنيميا المنجلية او الثلاسيميا ايضا مصابون
بمرض انيميا الفول.
يعرف المرض بين الأطباء بمرض نقص خميرة (انزيم) ديهيدروجينيز الجلوكوز 6
فوسفاتي(GLOCUSE 6 PHOSPHATE DEHYDROGENASE )او بالمختصر ( G6PD). يعبتر
هذا المرض مرض وراثي نتيجة لطفرة موجودة على كروموسوم اكس فلذلك يعتبر من
الأمراض الوراثية التي تنتقل بالوراثة المرتبطة باالجنس.وهو في العادة يصيب
الذكور و ينتقل من امهاتهم .وفي بعض الأحيان قد يظهر المرض على الاناث كما
ان الذكور المصابون بالمرض ينقلون المرض ولكنهم ينقلونه الى بناتهم ولا
ينقلنهوا الا ابنائهم مطلقا.ونقص اللأنزيم يجعل الكريات الدم الحمراء معرضه
للتحلل والتكسر قبل موعدها المعتاد(والذي في العادة يتجاوز 100 يوم) فيؤدي
الى انخفاض في الهيموجلوبين (فقر دم او انيميا)مع انتشار للمادة الصفراء
تعجز عن تصفيتة الكبد بشكل سريع.هناك تفاوت كبير في السن الذي تظهر فيه
اعراض المرض.فقد يظهر عند المواليد على مباشرة بعد الولادة فيكون اليرقان
عندهم اعلى من المستوى المعتاد و الذي يصيب الكثير من الأطفال الطبيعين كما
انه قد يحدث في أي سن و لكنه في العادة يظهر عند ما يتناول المصاب بالمرض
الفول او العدس او أي نوع من البقوليات او بعد الأصابة بمرض فيروسي او عند
تناول بعض من العقاقير.كما قد تظهر الاعراض من دون ان يصاب الشخص بأي مرض و
من دون ان يناول أي نوع من المواد المؤكسدة كلبقوليات.
الاعــــراض :
ليس هناك اعراض خاصة بمرض انيميا الفول.ولذلك تشترك اعراض هذا المرض
بالعديد من الأمراض التي تسبب تكسر او تحلل في في كريات الدم اللحمراء.
وهذه الاعراض هما فقر الدم(انيميا) و اليرقان(اصفرار الجلد و االعينين )وقد
يحدث تكسر في كريات الدم الحمراء بشكل حاد (مفاجئ) او بشكل مزمن
(ببطء).ولذلك يمكن ان نقسم اعراض المرض الى هذين القسمين
التكسر الحاد :
يسبب تكسر الكريات الحمراء الى انخفاض نسبة الهيموجلوبين في الدم.ولذلك
فالتعريف العلمي لفقر الدم او الأنيميا هو انخفاض مستوى الهيموجلوبين الى
مستوى اقل من المعدل الطبيعي (المعدل الطبيعي يختلف حسب العمر و الجنس
ولكن هو دائما فوق 10 غرامات لكل 100 مليمتر من الدم).و يكون لون الشخص
المصاب بفقر الدم شاحب و يستدل الطبيب الى ذلك بفحص الغشاء المبطن للجفن
السفلي للعين.فكلما قل الون الأحمر في الجفن كلما دل على انخفاض في مستوى
الهيموجلوبين .و عند تكسر كريات الدم الحمراء تنتنج مادة صفراء تعرف
بالبيلوروبين ( Bilirubin) وتيتخلص منها الجسم عن طريق الكبد.ولكن اذا
ارتفع مستوى البيلوربين في الدم(نتسجة لتكسر عدد كبير من كريات الدم
الحمراء دفعة واحدة) فانه يرشح والى الجلد وبقيت الاعضاء ويظهر لون الجلد و
العينين اصفر.و ارتفاع مادة البيلوربين في العادة ليست مضرها الا في
المواليد نظرا لضعف الجدار العازل لشرايين في المخ في ذلك العمر.ولذلك يجب
اخذ الأمر بمحمل من الحرص و الأهتمام في حالة اصفرار جلد حديثي الولادة.
في بعض انواع نقص انزيم G6PD يكون تكسر او تحلل كريات الدم الحمراء بطيء و
لا تتكسر الكريات بشكل مفاجىء.هذا النوع من التكسر يسبب فقر دم مزمن وقد
يكون مصحوب باصفرار بسيط في الجلد.و قد لا يحدث فقر دم اذا استطاع الجسم
انتاج كمية اعلى من الهيموجلوبين تعوض عن النقص المستمر.يحتاج الشخص المصاب
بهذا النوع من التكسر تغذية جيدة خاصة من فيتامين حمض الفوليك والذي يمكن
تناولة على شكل اقراص او شراب اذا لزم الامر.
في بعض الأحيان يحدث التكسر لأسباب مجهولة ولكن بشكل عام ان تعرض الجسم لأي
مادة مؤكسدة يمكن ان تكسر الدم. واليك بعض من اهم المواد المكسرة للدم
والتي ينصح بتجنبها :
1- تناول بعض الاطعمة:وهذي الأطعمة هي البقوليات بجميع انواعها خاصة
الفول و العدس و البازلاء والفاصوليا.و تتراوح كمية المادة المؤكسدة بين
نوع و اخر من الأطعمة. وقد كون الكمية التي يتناولها الشخص قليلة فلا تسبب
له مشكلة ولكن في الكثير من الأحيان يتناول الشخص كمية قليلة فتسبب له تكسر
حاد في الدم وا حياننا يتناول نوع معين من الأطعمة لسنوات عديدة ولا تسبب
له تكسر وفجاءة تتكسر لديه الكريات بعد تناول كمية قليلة منه.و نظرا للحاجة
لنقل الدم لبعض الأشخاص الذين انخفض لديهم مستوى الهيموجلوبين بشكل حاد
،فإننا ننصح جميع المصابين بمرض انيميا الفول تجنب جميع الاطعمة المتعارف
انها تسبب تكسر في الدم وعدم التهاون في هذا الأمر نظرا للمخاطر التي قد
يتعرض لها الشخص عند نقل الدم له خاصة من الأمراض المجهولة .
2- تناول بعض انواع من الأدوية:على كل المصابين بهذا المرض تنبية الطبيب
المعالج على انهم مصابون بهذا المرض لكي يتفادة اعطائهم بعض انواع من
الأدوية و استبدالها بانواع مأمونه،الا ذا كان الشخص مصاب بالملاريا و يضطر
الطبيب على استعمال دواء محدد لهذا المرض.(يمكن الاطلاع على هذه الأدويه
في صفحة العلاج).
3- التعرض للالتهابات الفيروسية او البكتيرية.
الاسبـــــــــاب :
ينتج انزيم G6PD جين موجود على كروموسوم أكس يعرف بجين انزيم G6PD.و حين
يتعطل هذا الجين يحدث مرض انيميا الفول.ولذلك يعتبر هذا المرض مرضاً من
امراض الجينات الوراثية.
يحدث في داخل الخلية الحمراء عدة تفاعلات كيميائية و على شكل شبكة مترابطة
من المواد الكيميائية تتفاعل مع بعضها البعض و مدعومة بالعديد من
الأنزيمات.و لو ركزنا على احد هذه الشبكات و المسمية التفاعل فسفات
البنتوز.ودور انزيم G6PD هو انتاج مادة البنتوز من جلوكوز الفسفات السداسي و
تحويل مادة النيكوتونوميد ادنين للفسفات الثنائي النووي (NADP ) الى مادة ا
لنيكوتونوميد ادنين للفسفات الثنائي النووي المؤكسد( NADPH).يوجد عدة طرق
اخرى لأنتاج مادة ( NADPH) في جميع خلايا الجسم ماعدى كريات الدم
الحمراء،فتفاعل فسفات البنتوز هي الطريقة الوحيد لأنتاج ( NADPH). يكمن
اهمية مادة ( NADPH)في انه يحافظ على جعل مادة الجلتثيون في داخل كرية الدم
الحمراء في حالة مختزلة قابلة لسحب الهيدروجين من أي مادة مؤكسدة كلفول
والبقوليات و بعض العقاقير والألتهابات وحماية الكرية الدم الحمراء من
التكسر.
وظيفة انزيم G6PD:
عند نقص انزيم G6PD يؤدي لنقص تكون مادة ( NADPH) ويجعل مادة الجلتثيون في
حالة مؤكسدة(حالة الأكسدة ضد الاختزال).الا هذا الحد لا تتأثر الكريات الدم
الحمراء بهذا النقص،ولكن عندما يأكل الشخص مادة الفول فان الهيموجلوبين في
داخل الخلية الحمراء يتبلور نتيجة لعدم حمايتة من قبل مادة الجلتثيون و
بذلك تكسر الكريات الدم الحمراء مؤديتا الى فقر دم وصفار.
الوراثة و انزيم G6PD :
لا شك ان مرض نقص انزيم G6PD يعتبر مرضاً وراثية ولكنه في العادة يظهر على
الذكور ولا يظهر على الاناث .و لذي ينقل المرض الى الذكور هن الأناث و
الذكور المصابة بالمرض تنقلة فقط لاناث.قد يبدوا ان الموضوع محير او معقد
ولكن يمكن فك كل الطلاسم بمعرفة الجانب الوراثي من القصة.ولكي يسهل عليك
فهم الموضوع عليك ان تعرف بعض اساسيات الوراثة .فمن المهم ان تعرف ما معنى
الخلية والنواة و الكروموسوم والجين. ولذلك عليك الذهاب الى هذا الرابط
للتعرف على كل ذلك ثم الرجوع لتكملة قراءة الموضوع.
يحدث مرض نقص انزيم G6PD بسبب وجود طفرة على الجين المنتج لذلك الانزيم
والموجود على الذراع الطويلة كروموسوم أكس.هناك انواع كثيرة من الطفرات
المؤدية لنقص او ضعف انزيم G6PD .ويعتقد انه يوجد فوق 400 نوع من
الطفرات.واختلاف انواع الطفرات يفسر الى حد ما الاختلاف في الأعراض،و هناك
بعض الطفرات التي تتميز بإحداث تكسر تلقائي لكريات الدم الحمراء للمواليد
وهناك انواع اخرى تتميز بحدوث تكسر تلقائي مستمر للدم حتى من دون التعرض
للمواد المؤكسدة كالفول..
يقع انزيم G6PD في الشريط رقم 28 من الذراع الطويلة من كروموسوم أكس.وبما
ان المرأة لديها نسختان من كروموسوم أكس فأنها لديها أيضا نسختان من جين
إنزيم G6PD نسخة أتتها من أمها و الأخرى أتتها من أبوها.وبما أن الرجل ليس
لدية الا نسخة واحدة من كروموسوم أكس، فأنه لا يحمل الا نسخة وحيدة من جين
إنزيم G6PD.و بما أن الجينات قابلة للعطب فأن حدوث عطب في احد نسختي جين
إنزيم G6PD في المرأة لا يؤدي في العادة للإصابة بالمرض،لان المرأة لديها
نسخة إضافية على كروموسوم أكس الأخر.ولكن الرجل على خطر،فأن إصابة نسختة
الوحيدة بعطب يسبب له المرض.
قسم الأطباء الأمراض الوراثية الا أربعة أقسام رئيسية .القسم الأول الأمراض
الوراثية المتعلقة بالكروموسومات،القسم الثاني الأمراض المتعلقة
بالجينات(ويسمى أيضا بأمراض وحيد المورثة)والنوع الثالث الأمراض الوراثية
المتعددة الأسباب ،والنوع الرابع الأمراض المتعلقة بالميتوكندريا.
و يتفرع من القسم الثاني أربع أنواع من الأنماط (الطرق) الوراثية التي
ينتقل فيها المرض من شخص إلى أخر.النمط الأول يسمى الوراثة المتنحية
،الثاني الوراثة السائدة،الثالثة الوراثة المتعلقة بالجنس
المتنحية،والرابعة الوراثة المرتبطة بالجنس السائدة.يعتبر مرض انزيم G6PD
من الأمراض المتعلقة بالجينات و ينتقل بالوراثة المتعلقة بالجنس المتنحية.و
سمي بالوراثة المتعلقة بالجنس المتنحية،لان الجين المعطوب يقع على إحدى
الكروموسومات المحددة للجنس ،كروموسوم أكس.و سمي بالمتنحى لأن المرض لا
يظهر على الشخص الا اذا أصيبت كل نسخ كروموسوم أكس بالعطب.ولذلك في العادة
لا يظهر على المرأة لآن لديها نسختان من كروموسوم أكس،ولكنه يظهر على الرجل
لآنه لا يحمل الا نسخة واحدة.ولمعرفة المزيد عن الوراثة المتعلقة بالجنس
المتنحية يمكنك الرجوع لهذه الصفحة:الوراثة المرتبطة بالجنس المتنحية.
متى يصاب كلا من الرجل و المرأة للمرض و متى تصبح المرأة حاملة للمرض ؟؟
يصاب الرجل بالمرض اذا كانت النسخة التي لدية من جين انزيم G6PD معطوب(به
طفرة).و بما ان جين انزيم انزيم G6PD موجود على كروموسوم أكس و الرجل ليس
لدية الا نسخة واحدة جاءته من امة فان المرض ينقل من الام لأبنائها . و
ابنائها المصابون لا ينقلون المرض لأبنائهم لانهم لا يعطون ابنائهم كرموسوم
أكس فهم يعطونهم كروموسوم واي .فلذلك لا ينتقل المرض من الرجل للأبنائه
الذكور اطلاقاً.ولكن الرجل من المصاب من الممكن ان ينقل المرض لبناته لانه
يعطيهن نسختة المعطوبة من كروموسوم أكس الورحيد عنده.(لتعرف على الفرق بين
الانثى والذكرمن ناحية التركيبة الوراثية ارجع لهذا الرابط: تحديد الجنس).
تكون المرأة حاملة للمرض اذا كانت فقط احدى نسختي الجين الموجودة على كروموسوم أكس مصابة بالعطب.
متى تحمل المرأة المرض
تكون المرأة حاملة للمرض اذا كانت فقط احدى نسختي الجين الموجودة على كروموسوم أكس مصابة بالعطب.
تصاب المرأة بالمرض اذا اصيبت كلتا النسختين التي لديها بالعطب.فلذلك نجد
ان النسختان التي على كروموسوم أكس من الاب ومن الام معطوبة.و هذا يعني ان
ابوها ايضا مصاب بالمرض و انه على اقل تقدير ان امها حاملة او مصابة
بالمرض.اذا فهمنا هذا الأمر كقاعدة فعلينا ان نعلم ان هناك حالات استثنائية
تصاب فيها المرأة بالمرض بدون ان تكون الأم حاملة او مصابة بالمرض وقد
يحدث احياناً ان الأم فقط حاملة للمرض او مصابة بينما الأب سليم.وفي ما يلي
الحالات التي تصاب بها المرأة بالمرض:
1-عندما يكون الأب مصاب والام حاملة او مصابة بالمرض.وتكثر هذه الحالة في
المجتمعات التي يكثر فيها زواج الأقارب ،خاصة الزواج بين ابناء الخالات او
الأخوال.
2- عندما يكون الأب فقط مصاب بالمرض بينما الأم سليمة(أي ليست حاملة و لا مصابة بالمرض).
واحدة منهما عليه نسخة من جين انزيم G6PD .و لذلك فالمرأة اذا عطبت احدى
نسختيها فانها في العادة لا تصاب المرض لان لديها نسخة اخرى.ولكن هناك امر
مهم يتحتم علينا معرفته من ناحية الفروقات الوراثية بين الرجل و المرأة.كما
نعلم ان الأنسان لدية 46 كروموسوم وهي تأتي على شكل ازواج،وكل نسخة من هذه
الأزواج تأتي من الأم والأب.ولو نظرنا الى الزوجد الجنسي لوجدنا ان المرأة
لديها نسختان من كروموسوم أكس بينما الرجل لديه فقط نسخة من كروموسوم أكس
واخرى من كروموسوم واي.و مع ان المرأة لديها نسختين من كروموسوم أكس في كل
خلية الا ان كل خلية لا يعمل فيها الا نسخة واحدة فقط و تقوم الخلية بتعطيل
النسخة الثانية و ذلك لعدم حاجتها
لها،و هذا شىء متوقع لان جميع خلايا الرجل تعمل بشكل طبيعي بنسخة واحدة
لذلك فالله عز وجل جعل الخلية الأنثوية تعطل الكوموسوم الزائد لديها ولذلك
لكي لا ينتج كميه مضاعفة من البروتينات.تسمى خاصية تعطيل كروموسوم أكس في
الخلية الأنثوية بتثبيط او تعطيل أكس(X inactivation ).و بما ان كل خلية
فيها كوموسوم أكس من الأب و اخر من الأم فان الخلية لا تفرق بينهما فهي
تقوم بتعطيل الكروموسوم بغض النظر عن مصدرة.هذه الخاصية تسمى تعطيل أكس
العشوائي( Random X in activation).ولذلك لو نظرنا الا جميع خلايا المرأة
لوجدنا ان بعض خلاياها يعمل بها كروموسوم أكس من الأب والبعض يعمل بها
كروموسوم من الأم.
و بإمكان الطبيب او اخصائي المختبر ان يشاهد كروموسوم أكس المعطل ملتصق
بجدار الخلية من الداخل ويظهر في خلايا الدم البيضاء على شكل عصيه صغيرة
معروفة بعصى الطبل( Drum stick)وفي الخلايا المبطنة للفم بأجسام بار ( Bar
Bodies).وبما ان التثبيط يتم بشكل عشوائي في جميع الخلايا،فاننا نجد ان
حوالي 50% من الخلايا في جسم المرأة يعمل بها كوموسوم أكس مصدرة الأب و 50%
يعمل بها كروموسوم أكس مصدرة الأم.طبعا لا يفرق عند الخلية اذا ماكان
الكروموسوم النشط او المعطل هو كروموسوم كان مصدرة الأم او الأب لان
الجينات الموجودة على كروموسوم أكس الأنثوي او الذكري متطابقة و لا حتى
يمكن معرفة الفرق بينهما من الناحية التكوينية.ولكن الأمر مهم اذا كان احدى
هذه الكروموسومات يحمل جيناً غير سليم او به طفرة.و أليك التفاصيل.
لو قلنا ان امرأة ما اباها مصاب بمرض انيميا الفول(أي ان كروموسوم أكس الذي
لدية به جين به طفرة سببت المرض)، وان امها سليمة(أي ان كلا نسختي
كروموسوم أكس التي لديها سليمة من العطب).و على الأب و الأم ان يعطيا نسخة
من كروموسوم أكس(و هذا شرط ليكون لطفل انثى لا ذكر)وبما ان الأم لديها
نسختان من أكس وكلتاهما سليمتان فان النسخة التي سوف تعطيها لابنتها
سليمة،بينما الأب فليس لدية الا نسخة و حيدة من كروموسوم أكس وعليها جين(
لإنزيم G6PD) معطوب فلذلك ليس لديه أي اختيار الا اعطائها اياه.طبعا المرأة
تعطي كروموسوماتها على شكل بويضة بها 23 كروموسوم ،و الرجل ايضاً يعطي
كروموسوماته على شكل حيوان منوي به 23 كروموسوم.وعند اتحاد البويضة
والحيوان المنوي تكتمل عدد الكروموسومات فتصبح 46 كروموسوم وهذه الخلية
تنقسم الى بلايين الخلايا لتكون انسان كامل.لو نظرنا الا خلايا هذه البنت
لوجدنا ان جميع الخلايا بها كروموسوم أكس عليه جين انزيم G6PD سليم(اتى من
الأم) و اخر عليه جين انزيم G6PD معطوب،ولذلك تكون هذه البنت حاملة للمرض
وليست مصابة وذلك نظراً لان الجين السليم ينتج كمية كافية من انزيم G6PD
ويغطي النقص الذي احدثة الجين المعطوب.الا هنا و الأمر طبيعي وهذا الذي
يحدث في كثير من الحالات. لو نظرنا الا جميع خلايا هذه البنت لوجدنا ان 50%
من خلاياها يعمل بها كروموسوم أكس السليم(الذي اتاها من امها)بينما البقية
يعمل بها كروموسوم أكس غير السليم(الذي اتاها من ابوها).هذا هو المعتادـ.و
لكن بما ان تعطيل (تثبيط)كروموسوم أكس يتم بشكل عشوائي ،أي ان كل خلية ليس
لها علاقة بالخلية الاخرى و اذا تعطل كروموسوم أكس من الأم في خلية ما
فليس من الضرورة ان الخلية الاخرى ان يتعطل كروموسوم أكس من الأب ،فأنه ليس
من الضرورة ان تكون نسبة كروموسومات أكس من الأم ومن الأب 50% لكل
منهما.بل من الممكن ان تكون مثلا 70% من الخلايا يعمل بها كروموسوم أكس
الذي من الأم، وقد يكون اكثر من ذلك او اقل.
لا يهمنا هنا نسبة الخلايا التي يعمل بها كروموسوم أكس الأتي من الأم(أكس
السليم)بقدر معرفة نسبة الخلايا التي يعمل بها كروموسوم أكس الأتي من
الأب(أكس الغير سليم)فلكما زادة نسبة الخلايا التي يعمل بها كروموسوم أكس
الأتي من الأب لكما زاد عدد الخلايا التي لا يعمل بها جين انزيم G6PD
المعطوب.وكلما زادة عدد الخلايا في الجسم والتي يعمل بها كروموسوم أكس
المعطوب كلما زاد احتمال ان تظهر اعراض مرض نقص انزيم G6PD على هذه البنت
حتى وان لم يكن جميع خلاياها تعاني من هذا النقص، والمهم هوا النسبة بين
عدد الخلايا التي يعمل بها جين انزيم G6PD و بين الخلايا التي لا يعمل
بها.وكلما زادة النسبة لصالح الجين الذي لا يعمل كلما زادة نسبة احتمال ان
تصاب البنت بالمرض ويظهر عليها اعراضة.ان هذا الشرح المطول هو شرح لما
يعرف بظاهرة ليون(Lionization ) او تعطيل كروموسوم أكس الغير عشوائي( Non
Random X inactivation).والشرح السابق ينتبق ايضا على الحالات التي يكون
فيها الأب سليم(كروموسوم أكس الذي يحملة عليه جين انزيم G6PD سليم)والأم
حاملة او مصابة بالمرض.ففي كل مره تحمل فيها الأم بانثى فان زوجها سوف يعطي
دائما بنسخة سليمة من كروموسوم أكس.بينما الأم من اللمكن ان تعطي نسخة
سليمة او معطوبة(هذا اذا كانت حاملة للمرض)او دائما تعطي بنسخة معطوبة(لو
كانت مصابة بالمرض وكلتا نسختيها من كروموسوم أكس معطوبتان).
3- عندما تكون البنت مصابة بمرض أخر يسمى بمتلازمة تيرنر.فهذا المرض يصيب
البنات فقط وهو ناتج عن نقص في عدد الكروموسومات .فبدل ان يكون مجموع
الكروموسومات 46 يكون لديها فقط 45 كروموسوم .والناقص هو احد نسختي
كروموسوم أكس.فإذا حدث وكانت النسخة التي لديها فيه اجين انزيم G6PD معطوب
فأنها تصاب بالمرض وتشابه حالتها حالة الذكر المصاب بالمرض فهو لا يملك الا
نسخة واحدة من كروموسوم اكس وبه نسخة معطوبة من جين انزيم G6PD.
احتمالات الاصابة :
تعتمد احتمالات تكرر المرض في العائلة الواحدة حسب الحالة المرضية
للوالدين.ونظراً الا ان نقص الانزيم لا يظهر على الأناث فاننا سوف نعتمد
على الحالة الجينية للأنزيم مباشرة.و لكن على القارئ ان يعرف ان الأطباء في
العادة يقيسون كمية الانزيم في الدم و لا يجرون تحليل للجين مباشرة.و
السبب في ذلك هو صعوبة اجراء الفحص الجيني على المرضى و لا يجرى هذا
التحليل الى في الدراسات و الأبحاث الطبية
فيما يلي بعض الحالات الجينية للوالدين واحتمالات الأصابة:
الام سليمة و الزوج مصاب /
انظراً لوجود جين انزيم G6PD على كروموسوم أكس وليس على كروموسوم واي،و
نظراً الى ان الأب يعطي ابنائة الذكور كروموسوم واي ،فإن جميع ابنائة
الذكور يكونوا سليمين من المرض.بينما جميع بناتة حاملات للمرض.طبعا المرأة
الحاملة للمرض لا تصاب بالمرض و لا تظهر عليها اعراض المرض،ولكن عليك ان
تعرف انه في بعض الأحيان تظهر اعراض المرض على المرأة الحاملة للمرض،ولذلك
عليك الرجوع الى الفقرة التي تتحدث عن الحالات التي تصاب بها المرأة في
اعلى هذه الصفحة.انظر جدول تلقيح البويضات والحيوانات المنوية للأم
السليمة والزوج المصاب.
الام حاملة للمرض و الزوج سليم /
ان احتمال اصابة الذكور في هذه الحالة 50% في كل مرة تحمل فيها تحمل فيها
الأم.كذلك 50% من الأناث يكن حاملات للمرض كأمهن وفي العادة لا يصبن
بالمرض(ولكن ارجع للحالات الأستثنائية لاصابة المرأة في اعلى الصفحة). انظر
جدول تلقيح البويضات والحيوانات المنوية للأم الحاملة للمرض والزوج سليم.
الام حاملة للمرض و الزوج مصاب /
ان احتمال اصابة الذكور في هذه الحالة لا ترتفع عن الحالة السابقة فهي 50%
في كل حمل.ولكن احتمال اصابة الاناث في هذه الحالة تصل الى 25% ( أي ان كلا
نسختي جين انزيم G6PD معطوبتان)و الباقي منهن حاملات للمرض(أي لديهن نسخة
واحدة من جين انزيم G6PD معطوب و نسخة سليمة). انظر جدول تلقيح البويضات
والحيوانات المنوية للأم الحاملة للمرض والزوج المصاب.
الام مصابة و الزوج سليم /
ان احتمال اصابة الذكور في هذه الحالة هي 100% كما ان 100% من الاناث
حاملات للمرض. انظر جدول تلقيح البويضات والحيوانات المنوية للأم المصابة
بالمرض والزوج السليم.
الام و الزوج كلاهما مصاب /
و الأم في هذه الحالة مصابة بالمرض و كلا نسختي الجين من انزيم G6PD
معطوبتين .ان احتمال اصابة الذكور والأناث في هذه الحالة 100%. انظر جدول
تلقيح البويضات والحيوانات المنوية للأم و الاب المصابين.
العـــــــــلاج :
لا يوجد للأسف علاج شافي من هذا المرض،و لا يمكن التخلص منه ومنعه من
الانتقال من جيل لاخر.المرض ليس بالمعدي ولي لمخالطي المصاب او حاملي المرض
أي خطر على الأخرين .و المرض لا ينتقل بالمعاشرة الجنسية ولكن كما هو
معروف فالصفات الوراثية و يدخل في ذلك الجينات تنتقل من اتحاد الحيوان
المنوي بالبويضة .
العلاج من هذا المرض يتمحور حول تجنب تكسر الدم عن طريق تجنب التعرض للمواد
المؤكسدة كانواع معينة من الاطعمة والادوية و الالتهابات بشكل عام
الأطعمة التي يجب تجنبها
جميع البقوليات
الأدوية التي يجب تجنبها
Vitamains الفيتامينات
Vitamin K
Ascorbic acid(Vitamin C)
Anti-Malarial Drugs مضادات الملاريا
Pamaquine
chloroquine
Hydroxychloroquine
Mepacrine (quinacrine)
pamaquine
primaquine
Pentaquine
Quinocide
Quinocide
Quinaorine (Atabrine),
Quinine (this is used sometimes in soft drinks such as tonic water)Biguanides
Pyrimidines
Quinolines
Sulphonamides
Sulphones
Sulfanamides/ Sulfones مضادات السلفا
Sulfacetamide
Sulfanilamide
Salioylazulfapyridine (Azulfidine)
Sulfapyridine
Sulfasalazine
Sulfamethoxypyrimidine
Sulfaoethamide
N2 – Adetylbulfanilamide
2-Amino-5-Sulfanilylthiazole
Sulfiboxazole (Gantrisin)
Thizolsulfone (Promizole)
Diaminodipenysulfone (DDS)
Solfoxone (or Sulphoxone; Dapsone)
Anti-Inflammatory / Analgesic Drugsالمسكنات و مضادات الروماتزم
Salicylic Acid (Aspirin)
Phenacetin
Antipyrine
Aminopyrine (Pyramidon)
Aniline
Fenamic Acid
Pyrazolone (Acetanilide)
para-aminosalicylic acid
Anti-Bacterial Drugs المضادات الحيوية
co-trimoxazole
Cephalsoporins
Chloramphenicol
Nitrofurans (Furadentin)
Nalidixic acid
Furaltadone (Altafur)
Nitrofurazone (Furadin)
Penicillins
Quinolones
Chloramphenicol
Anti-Emetic / Neuroleptic Drugs مضادات التقيؤ
Phenothiazine derivatives
Uricosuric Drugs طاردت ليوريك
Benzoic Acid derivatives
Anti-Histaminic Drugsمضادات الهستامين
Antazoline
Cardiac/Anti-Hypertensive Drugs ادوية القلب و الضفط
Procainamide
Quinidine
Hydralazine
Anti-Tuberculous Drugs مضادات السل
Isoniazide
P.A.S.
Anti-Protozoal Drugsمضادت الطفيليات
Naphthalene
Miscellaneous متفرقات
Chelating Agents
Dimercaprol
Methylene blue
Naphthalene
Doxorubicin
L-dopa
انواع محددة من الأعشاب الصينية
عدل سابقا من قبل أحمد في الخميس يناير 27, 2011 11:57 pm عدل 1 مرات
DREAM MAN- مدير موقع المعهد الطبي
- تاريخ التسجيل : 25/01/2010
عدد المساهمات : 533
المزاج : رايق
الجنس :
رد: امراض الدم الوراثية بالتفصيل و بالعربي
من طرف DREAM MAN الخميس يناير 27, 2011 11:49 pm
الهيموفيليــــــــا ( مرض الناعور ) :
تخثر الدم
ما هو تخثر الدم؟
تخثر الدم هو ردة فعل الجسم على إصابة الأوعية الدموية والنزيف، وهو يشمل
جهداً متناسقاً بين الصفيحات وعدد كبير من بروتينات التخثُّر في الدم (أو
العوامل) من ضمن ذلك " العامل النسيجي" الذي يؤدي إلى حدوث تجلط الدم.
العامل النسيجي هو عبارة عن بروتين يتعرّض للدم عندما يتعرض وعاء دموي ما
إلى إصابة ( تسبب النزيف). وتبدأ عملية تخثر الدم عندما يتّحد العامل
النسيجي مع نوع معيّن من بروتين التخثّر يسمى العامل السابع المفعل(FVIIa) .
وبالتالي يشكّل الاتحاد بين العامل النسيجي والعامل السابع المفعل هو
الخطوة الأولى في عملية تؤدي في الواقع إلى تشكل خثرة دموية قوية ومستقرة
من شأنها أن توقف وتمنع أي نزيف آخر.
و بشكل عام، تتم السيطرة على النزيف بسرعة كبيرة من دون الحاجة إلى تدخل
طبي عند الأشخاص الأصحاء. وفي حالات الرض الشديد أو العملية الجراحية، على
الأطباء مساعدة المرضى على تحقيق تخثر دموي كافي – بهدف تقليص خسارة الدم
والإصابات المتعلقة بها.
غير أنّ بعض الأشخاص يولدون وهم مصابون باضطراب نزفي خلقي يؤدي إلى تدهور
قدرتهم على تكوين خثرة دموية، وتكون أغلب هذه الاضطرابات النزفية وراثية أو
شائعة في العائلة، مثال على ذلك الهيموفيليا (الناعور).
كما أنّ بعض الأشخاص الذين لم يعانوا قط من مشاكل نزفية يمكن أن يصابوا
بحالة تسبّب لهم النزيف تٌعرف بـ "الهيموفيليا المكتسبة". وفي هذا النوع من
الاضطرابات، تتطلب أصغر الجروح والكدمات تدخلا ً طبياً.و تشكل الهيموفيليا
و الأمراض المشابهة لها عبئاً طبياً و اقتصادياً ثقيلاً على كاهل المرضى و
المجتمع بشكل عام.
ما هو مرض الهيموفيليا؟
الهيموفيليا أو مرض الناعور هو عبارة عن اضراب نزفي خلقي يصيب
الذكورونادراً جداً النساء. ويصيب الهيموفيليا نوع "أ" و"ب" حوالى ٢٠٠٠٠٠
إلى ٣٠٠٠٠٠ ألفشخص في العالم.
يعاني المرضى المصابون بالهيموفيليا نوع "أ" من نقص جزئي أو تام أو خلل في
إنتاج بروتين التخثّر في الدم أو العامل FVIII، أما المصابون بالهيموفيليا
نوع "ب" فيعانون من نفس المشكلة مع العامل (FIX). وتكون درجة الهيموفيليا
"شديدة" عندما تقل فعالية عامل التخثّر المصاب (FVIII أو FIX) عن ١% من
المعدل الطبيعي. في حين أنّ الهيموفيليا "الخفيفة" تكون عندما يكون معدل
فعالية عامل التخثّر المتأثر أكبر من ٢٥% لكن أقل من المعدل الطبيعي. أما
الهيموفيليا "المعتدلة" فتكون عندما تتراوح فعالية عامل التخثّر بين ١-٥ %
من المعدل الطبيعي. ويعاني حوالى ٥٠ % من المرضى من الهيموفيليا الشديدة
الذين يحتاجون للمعالجة من النزيف مرات عديدة في الشهر.
وتظهر أعراض الهيموفيليا الشديدة عادةً في السنة الأولى من حياة الشخص
المصاب – وغالباً بمجرد أن يبدأ الطفل بالحركة. ويحدث النزف عادة ًفي
المفاصل (لاسيما في مفاصل التي تتحمل الوزن كالركبة والكاحل) وقد يؤدي
المرض إذا لم تتم معالجته إلى آلام مبرحة وتلف دائم وإعاقة. (وثمة نزيف
خفيف أو معتدل أو مهدّد بحدوث إعاقة قد يصيب العضلات أو الأنسجة الداخلية
أو القناة الهضمية أو حتى الدماغ). كما أنّ أي رض أو عملية جراحية بسيطة أو
كبرى أو خلع الأسنان عند المصابين بالهيموفيليا قد يتطلب تدخلاً طبياً
لمنع أو معالجة النزيف الذي قد ينشأ.
ما الذي يسبب مرض الهيموفيليا ؟
إن مرض الهيموفيليا هو مرض وراثي وهذا يعني أن هناك جينات لا تعمل بشكل
طبيعي هي التي تسبب هذا المرض. ومثل أي مشكلة صحية وراثية أخرى فإن
الهيموفيليا يمكن أن تنتقل من جيل إلى آخر، وفي كل الحالات تكون الجينات
تقريبا هي المسؤولة عن انتقال مرض الهيموفيليا من الأم إلى الأبناء أثناء
فترة الحمل .
ومع ذلك، فإن من بين كل 10 حالات هناك 3 حالات ولادة لطفل مصاب
بالهيموفيليا في عائلات ليس لديها تاريخ إصابة بهذا المرض ، وهناك 3 أسباب
لحدوث ذلك :
1- ربما يكون مرض الهيموفيليا موجود في العائلة لعدة أجيال ، ونظرا لعدم
ظهور علامات زيادة في نزيف الدم لدى الذكور فلا أحد يعرف أن مرض
الهيموفيليا موجود. وربما يوجد في العائلة فتيات يحملن مرض الهيموفيليا
ولكن إذا لم يكن من بين تلك الفتيات أي واحدة قد أنجبت أولادا ، أو ليس
هناك أي واحد من الأبناء كان مصاباً بالمرض فلن يعرف أي أحد بأن مرض
الهيموفيليا ينتقل بين هذه الأجيال حتى يولد طفل ذكر مصاب بهذا المرض.
2- قد تكون أمهات الأولاد قد حدثت لديهن طفرة جينية عند حدوث الحمل ، فالأم
هي الشخص الأول الذي يحمل المرض في العائلة وربما يحملن بناتها المرض أيضا
وربما يصاب أبناءها بهذا المرض.
3- ربما تكون الطفرة الجينيه التي تسبب الهيموفيليا قد وقعت عند حدوث الحمل
بأحد الأولاد. وفي هذه الحالة فإن الطفرة قد حدثت في بويضة الأم وانتقلت
إليه، وبالتالي فإن الأم في هذه الحالة ليست حاملة للمرض. لمزيد لمعلومات
عن الوراثة انظر فصل "كيف يصاب الأطفال بمرض الهيموفيليا".
ما هي الأسماء الأخرى للهيموفيليا- أ و الهيموفيليا- ب ؟
هيموفيليا- أ لها اسمان آخران هما :
• هيموفيليا تقليديه أوكلاسيكية لأنها هي الأكثر انتشارا من بين هيموفييليا نقص العوامل
• هيموفيليا نقص عامل 8 لأنها تحدث بسبب نقص بروتين عامل 8 في الدم الذي يسبب مشكلة في التخثر .
الهيموفيليا- ب أيضا لها اسمان هما :
• مرض كريستماس : سمي كذلك لان ستيفن كريستماس وهو كندي كان أول مريض تم تشخيص إصابته بهذا المرض بشكل مؤكد سنة 1952م
• هيموفيليا نقص عامل9، لأنها تحدث بسبب نقص عامل 9 في الدم والذي بفقدانه تصبح عملية التخثر الطبيعية بطيئة.
ما هو مدى انتشار مرض الهيموفيليا ؟
أن النوعين من هذا المرض (الهيموفيليا- أ والهيموفيلبيا- ب) كلاهما نادر
الحدوث ولله الحمد فمرض هيميوفيليا- أ يصاب به شخص واحد من بين 10000 شخص ،
أما الهيموفيليا- ب فهو أقل شيوعا، حيث يصاب تقريباً شخص واحد من بين 35000 شخص.
من الذي يصاب بمرض الهيموفيليا ؟
إن مرض الهيموفيليا يصيب الناس من كل الجنسيات والألوان والأصول العرقية حول العالم.
ومعظم أشكال الهيموفيليا الشديدة تصيب الذكور فقط ، أما إصابة الإناث
بالنوع الشديد من المرض فإنها تحدث فقط إذا كان الأب مصاباً بهذا المرض
والأم حاملة له وهذا شيء نادر الحدوث. ومع ذلك ، فإن العديد من النساء
الحاملات لهذا المرض تظهر عليهن أعراض طفيفة لمرض الهيموفيليا ، ونحن الآن
نعرف تماماً أن حاملات المرض يمكن أن يصبن بمشاكل النزيف وهذا يمكن أن يؤثر
على نوعية حياتهن.
وبما أن الهيموفيليا مرض وراثي فالأطفال يصابون به منذ لحظة الولادة، وفي
الحقيقة فإن مرض الهيموفيليا يتم تشخيصه في السنة الأولى من الحياة، إنها
مشكلة تستمر مدى الحياة، وحتى الآن لا توجد طريقة لتصحيح العيب الوراثي.
ما مدى خطورة مرض الهيموفيليا ؟
يمكن تقسيم هيموفيليا- أ وهيموفيليا- ب إلى 3 أنواع من ناحية الخطورة حسب الجدول التالي :-
نسبه الحالات المصابة ( 40% من حالات الهيموفيليا ) مستوى عامل 8 أو 9 في الدم * ( أقل من 1% ) تصنيف الحالات ( شديدة )
نسبه الحالات المصابة ( 20% - 25% من حالات الهيموفيليا ) مستوى عامل 8 أو 9
في الدم * ( 1- 5% من النسبة الطبيعية) تصنيف الحالات ( متوسطة)
نسبه الحالات المصابة ( 35% - 40% من حالات الهيموفيليا ) مستوى عامل 8 أو 9
في الدم * ( 5- 30% من النسبة الطبيعية ) تصنيف الحالات ( بسيطة )
*نشاط عامل التخثر لدى الشخص الطبيعي يعتبر 100% ويتراوح ما بين 50% -150%
المرضى المصابون بالهيموفيليا الشديدة يكون لديهم مستوى عامل 8 أو عامل 9
أقل من 1% من المستوى الطبيعي في الدم ، وبدون علاج وقائي يمكن أن يصابوا
بالنزف عدة مرات في الشهر، وليس هناك سبب واضح للنزيف ، وهذا يدعى النزيف
التلقائي.
والمصابون بالهيموفيليا متوسطة الدرجة عادة ينزفون بدرجة أقل في معظم وهذا
النزف يتكرر بسبب الإصابة برضة غير خطيرة مثل جروح الرياضة ، ومع ذلك فبعض
المصابين بالهيموفيليا متوسطة الدرجة خصوصا أولئك الذين لديهم مستوى عامل 9
أو عامل 8 حوالي 2% أو أقل يمكن أن يتكرر عندهم نزف الدم العفوي وبنفس
الطريقة التي تحدث عند الأشخاص المصابين بمرض الهيموفيليا الشديدة.
أما الأشخاص المصابين بالهيموفيليا البسيطة فإنهم يصابون أيضا بالنزيف ولكن
بدرجة أقل بكثير، وربما يدركون وجود مشكلة النزيف فقط في حالة إجراء جراحة
أو خلع سن أو إصابة خطيرة. والخطر بالنسبة للمرضى المصابين بالهيموفيليا
البسيطة يتمثل في أنهم يصابون بحالات نزيف قليلة جدا وبالتالي فهم عادة لا
يعرفون ماذا يفعلون عندما يحدث النزيف ، والنساء الحاملات لهذا المرض ربما
ينزفن أكثر خلال الدورة الشهرية ، ولهذه الأسباب فالأشخاص المصابون
بالهيموفيليا البسيطة هم أيضا بحاجة لمتابعة حالتهم في مركز علاج
الهيموفيليا. لمزيد من المعلومات أنظر الهيموفيليا البسيطة والمتوسطة
هل هناك علاج فعال لمرض الهيموفيليا ؟
نعم ، ولله الحمد ، يوجد علاج فعال، والأدويه الحالية لهيموفيليا- أ و
هيموفيليا- ب فعالة جدا ، وأساس العلاج لهذا المرض هو استخدام عامل التخثر
المركز حيث يتضمن هذا العلاج إدخال عامل التخثر المفقود إلى الدم عند
الأطفال المصابين بهذا المرض، وهي طريقة آمنة وفعالة لإيقاف النزيف ، وحتى
هذا العلاج يمكن أن يستعمل كطريقة وقائية لمنع النزيف من الحدوث كليا.
وفي هذه الأيام فإن الأطفال الذين يولدون مصابين بهذا المرض يمكن أن تنتظرهم حياة صحية ونشطة كغيرهم من الاطفال .
المضاعفات يمكن أن تحدث فعلا، ومن أخطرها ظهور المثبطات فبعض المصابين
بالهيموفيليا لا يستجيب جهازهم المناعي بشكل فعال لعامل التخثر المركز الذي
يتم تزويدهم به لوقف أو منع النزيف نتيجه لتكون المثبطات ، فالعامل المركز
يتعرف عليه الجسم كمادة غريبة عليه ولأن دفاعات الجسم لا تميزه فإن أجهزة
المناعة في الجسم تقاوم هذا الجسم الغريب بتكوين أجسام مضادة وهي مواد
طبيعية تسبح في الدم ، والأجسام المضادة التي تكونت في الجسم تبطل مفعول
العامل المركز المحقون في الوريد ومن ثم تمنعه من القيام بعمله أي وقف
النزيف، وهذه الأجسام المضادة تسمى المثبطات ، ولحسن الحظ هناك علاجات
فعالة للأطفال الذين تتكون لديهم المثبطات.
كيف يتخثر الدم في الوضع الطبيعي؟
ينتقل الدم في أنحاء الجسم داخل شبكة من الأوعية الدموية، وعندما تجرح
الأنسجة يحدث ضرر في الأوعية الدموية يسبب تسرب الدم من خلال ثقوب في جدران
تلك الأوعية ، وقد تنفتح أو تجرح الأوعية القريبة من سطح الجلد كما في
حالة جروح الجلد أو قد تجرح في أماكن عميقة داخل الجسم ما ينتج عنه كدمة أو
نزيف داخلي ، لمزيد من المعلومات وللتعرف على أنواع النزيف المختلفة أنظر
التعامل مع حالات نزف الدم .
أن الجلطة أو التخثر هي عملية معقدة تجعل من الممكن إيقاف نزف الأوعية
الدموية المجروحة وحالما تجرح جدران الأوعية الدموية تعمل البروتينات معا
لتكوين جلطة تشكل سدادة للجرح وهناك عدة خطوات تساهم في تكوين هذه السدادة
في مكان الإصابة.
والمتأمل في الخطوات والمراحل التاليه التي يتخذها الجسم في التعامل مع النزيف خير دليل على أعجاز الخالق سبحانه وتعالى
المرحلة الاولى : تتقلص الأوعية الدموية لتبطئ تدفق الدم للمنطقة المجروحة. وهذا يدعى انقباض الأوعية .
المرحلة الثانيه : الصفائح الدموية ، وهي عبارة عن قطع صغيرة جدا من
الخلايا تكون أول من يصل إلى مكان الإصابة حيث تلعب الصفائح دورا هاما في
عملية وقف النزيف عن طريق تجمعها معا وبذلك يبدأ إصلاح وعلاج الأوعية
الدموية المجروحة. وهذا يدعى التصاق الصفائح.
المرحلة الثالثه : تبعث هذه الصفائح إشارات كيميائية تدعوا فيها الصفائح
الدموية الأخرى وعوامل التخثر لمساعدتها مثل عامل ويلبراند، وهذه الصفائح
المنتشرة تطلق المواد التي تنشط الصفائح المجاورة الأخرى والتي تتجمع معا
في مكان الجرح وتشكل سدادة من الصفائح، وهذه العملية تسمى تجمع اوتكدس
الصفائح .
المرحلة الرابعه : يشكل سطح هذه الصفائح المنشطة مكانا ملائما لحدوث الجلطة
حيث تتحد مع عوامل التخثر الموجوده في تيار الدم لتكوين سلسلة تسمى
الفيبرين حيث تنتظم حزم الفيبرين معا لنسج شبكة حول الصفائح الدموية وهذا
يمنع الصفائح الدموية من الانزلاق والعودة في تيار الدم. وهذه البروتينات
اوالعوامل 1، 2، 5، 7،8، 9، 10،11، 13 تعمل مثل قطع الدومينو في التفاعل
المسلسل وهذه العملية تسمى حاجز التخثر.
ما هي مشكلة التخثر في حالة الإصابة بمرض الهيموفيليا ؟
عندما يُفقَد أحد البروتينات (على سبيل المثال عامل 8 ) فإن سلسلة التفاعل
تنقطع ولا يحدث التخثر، أو يحدث بصورة أبطأ بكثير من الوضع الطبيعي.
والصفائح الدموية في مكان الجرح لا تتشابك في مكان الإصابة لتكوين جلطة
دموية دائمة ، وتكون الجلطة طرية حيث تسهل إزاحتها من مكانها ، وبدون علاج
يمكن أن يستمر النزيف لعدة أيام وأحيانا عدة أسابيع ويتكرر عادة حدوث
النزيف.
تخثر الدم
ما هو تخثر الدم؟
تخثر الدم هو ردة فعل الجسم على إصابة الأوعية الدموية والنزيف، وهو يشمل
جهداً متناسقاً بين الصفيحات وعدد كبير من بروتينات التخثُّر في الدم (أو
العوامل) من ضمن ذلك " العامل النسيجي" الذي يؤدي إلى حدوث تجلط الدم.
العامل النسيجي هو عبارة عن بروتين يتعرّض للدم عندما يتعرض وعاء دموي ما
إلى إصابة ( تسبب النزيف). وتبدأ عملية تخثر الدم عندما يتّحد العامل
النسيجي مع نوع معيّن من بروتين التخثّر يسمى العامل السابع المفعل(FVIIa) .
وبالتالي يشكّل الاتحاد بين العامل النسيجي والعامل السابع المفعل هو
الخطوة الأولى في عملية تؤدي في الواقع إلى تشكل خثرة دموية قوية ومستقرة
من شأنها أن توقف وتمنع أي نزيف آخر.
و بشكل عام، تتم السيطرة على النزيف بسرعة كبيرة من دون الحاجة إلى تدخل
طبي عند الأشخاص الأصحاء. وفي حالات الرض الشديد أو العملية الجراحية، على
الأطباء مساعدة المرضى على تحقيق تخثر دموي كافي – بهدف تقليص خسارة الدم
والإصابات المتعلقة بها.
غير أنّ بعض الأشخاص يولدون وهم مصابون باضطراب نزفي خلقي يؤدي إلى تدهور
قدرتهم على تكوين خثرة دموية، وتكون أغلب هذه الاضطرابات النزفية وراثية أو
شائعة في العائلة، مثال على ذلك الهيموفيليا (الناعور).
كما أنّ بعض الأشخاص الذين لم يعانوا قط من مشاكل نزفية يمكن أن يصابوا
بحالة تسبّب لهم النزيف تٌعرف بـ "الهيموفيليا المكتسبة". وفي هذا النوع من
الاضطرابات، تتطلب أصغر الجروح والكدمات تدخلا ً طبياً.و تشكل الهيموفيليا
و الأمراض المشابهة لها عبئاً طبياً و اقتصادياً ثقيلاً على كاهل المرضى و
المجتمع بشكل عام.
ما هو مرض الهيموفيليا؟
الهيموفيليا أو مرض الناعور هو عبارة عن اضراب نزفي خلقي يصيب
الذكورونادراً جداً النساء. ويصيب الهيموفيليا نوع "أ" و"ب" حوالى ٢٠٠٠٠٠
إلى ٣٠٠٠٠٠ ألفشخص في العالم.
يعاني المرضى المصابون بالهيموفيليا نوع "أ" من نقص جزئي أو تام أو خلل في
إنتاج بروتين التخثّر في الدم أو العامل FVIII، أما المصابون بالهيموفيليا
نوع "ب" فيعانون من نفس المشكلة مع العامل (FIX). وتكون درجة الهيموفيليا
"شديدة" عندما تقل فعالية عامل التخثّر المصاب (FVIII أو FIX) عن ١% من
المعدل الطبيعي. في حين أنّ الهيموفيليا "الخفيفة" تكون عندما يكون معدل
فعالية عامل التخثّر المتأثر أكبر من ٢٥% لكن أقل من المعدل الطبيعي. أما
الهيموفيليا "المعتدلة" فتكون عندما تتراوح فعالية عامل التخثّر بين ١-٥ %
من المعدل الطبيعي. ويعاني حوالى ٥٠ % من المرضى من الهيموفيليا الشديدة
الذين يحتاجون للمعالجة من النزيف مرات عديدة في الشهر.
وتظهر أعراض الهيموفيليا الشديدة عادةً في السنة الأولى من حياة الشخص
المصاب – وغالباً بمجرد أن يبدأ الطفل بالحركة. ويحدث النزف عادة ًفي
المفاصل (لاسيما في مفاصل التي تتحمل الوزن كالركبة والكاحل) وقد يؤدي
المرض إذا لم تتم معالجته إلى آلام مبرحة وتلف دائم وإعاقة. (وثمة نزيف
خفيف أو معتدل أو مهدّد بحدوث إعاقة قد يصيب العضلات أو الأنسجة الداخلية
أو القناة الهضمية أو حتى الدماغ). كما أنّ أي رض أو عملية جراحية بسيطة أو
كبرى أو خلع الأسنان عند المصابين بالهيموفيليا قد يتطلب تدخلاً طبياً
لمنع أو معالجة النزيف الذي قد ينشأ.
ما الذي يسبب مرض الهيموفيليا ؟
إن مرض الهيموفيليا هو مرض وراثي وهذا يعني أن هناك جينات لا تعمل بشكل
طبيعي هي التي تسبب هذا المرض. ومثل أي مشكلة صحية وراثية أخرى فإن
الهيموفيليا يمكن أن تنتقل من جيل إلى آخر، وفي كل الحالات تكون الجينات
تقريبا هي المسؤولة عن انتقال مرض الهيموفيليا من الأم إلى الأبناء أثناء
فترة الحمل .
ومع ذلك، فإن من بين كل 10 حالات هناك 3 حالات ولادة لطفل مصاب
بالهيموفيليا في عائلات ليس لديها تاريخ إصابة بهذا المرض ، وهناك 3 أسباب
لحدوث ذلك :
1- ربما يكون مرض الهيموفيليا موجود في العائلة لعدة أجيال ، ونظرا لعدم
ظهور علامات زيادة في نزيف الدم لدى الذكور فلا أحد يعرف أن مرض
الهيموفيليا موجود. وربما يوجد في العائلة فتيات يحملن مرض الهيموفيليا
ولكن إذا لم يكن من بين تلك الفتيات أي واحدة قد أنجبت أولادا ، أو ليس
هناك أي واحد من الأبناء كان مصاباً بالمرض فلن يعرف أي أحد بأن مرض
الهيموفيليا ينتقل بين هذه الأجيال حتى يولد طفل ذكر مصاب بهذا المرض.
2- قد تكون أمهات الأولاد قد حدثت لديهن طفرة جينية عند حدوث الحمل ، فالأم
هي الشخص الأول الذي يحمل المرض في العائلة وربما يحملن بناتها المرض أيضا
وربما يصاب أبناءها بهذا المرض.
3- ربما تكون الطفرة الجينيه التي تسبب الهيموفيليا قد وقعت عند حدوث الحمل
بأحد الأولاد. وفي هذه الحالة فإن الطفرة قد حدثت في بويضة الأم وانتقلت
إليه، وبالتالي فإن الأم في هذه الحالة ليست حاملة للمرض. لمزيد لمعلومات
عن الوراثة انظر فصل "كيف يصاب الأطفال بمرض الهيموفيليا".
ما هي الأسماء الأخرى للهيموفيليا- أ و الهيموفيليا- ب ؟
هيموفيليا- أ لها اسمان آخران هما :
• هيموفيليا تقليديه أوكلاسيكية لأنها هي الأكثر انتشارا من بين هيموفييليا نقص العوامل
• هيموفيليا نقص عامل 8 لأنها تحدث بسبب نقص بروتين عامل 8 في الدم الذي يسبب مشكلة في التخثر .
الهيموفيليا- ب أيضا لها اسمان هما :
• مرض كريستماس : سمي كذلك لان ستيفن كريستماس وهو كندي كان أول مريض تم تشخيص إصابته بهذا المرض بشكل مؤكد سنة 1952م
• هيموفيليا نقص عامل9، لأنها تحدث بسبب نقص عامل 9 في الدم والذي بفقدانه تصبح عملية التخثر الطبيعية بطيئة.
ما هو مدى انتشار مرض الهيموفيليا ؟
أن النوعين من هذا المرض (الهيموفيليا- أ والهيموفيلبيا- ب) كلاهما نادر
الحدوث ولله الحمد فمرض هيميوفيليا- أ يصاب به شخص واحد من بين 10000 شخص ،
أما الهيموفيليا- ب فهو أقل شيوعا، حيث يصاب تقريباً شخص واحد من بين 35000 شخص.
من الذي يصاب بمرض الهيموفيليا ؟
إن مرض الهيموفيليا يصيب الناس من كل الجنسيات والألوان والأصول العرقية حول العالم.
ومعظم أشكال الهيموفيليا الشديدة تصيب الذكور فقط ، أما إصابة الإناث
بالنوع الشديد من المرض فإنها تحدث فقط إذا كان الأب مصاباً بهذا المرض
والأم حاملة له وهذا شيء نادر الحدوث. ومع ذلك ، فإن العديد من النساء
الحاملات لهذا المرض تظهر عليهن أعراض طفيفة لمرض الهيموفيليا ، ونحن الآن
نعرف تماماً أن حاملات المرض يمكن أن يصبن بمشاكل النزيف وهذا يمكن أن يؤثر
على نوعية حياتهن.
وبما أن الهيموفيليا مرض وراثي فالأطفال يصابون به منذ لحظة الولادة، وفي
الحقيقة فإن مرض الهيموفيليا يتم تشخيصه في السنة الأولى من الحياة، إنها
مشكلة تستمر مدى الحياة، وحتى الآن لا توجد طريقة لتصحيح العيب الوراثي.
ما مدى خطورة مرض الهيموفيليا ؟
يمكن تقسيم هيموفيليا- أ وهيموفيليا- ب إلى 3 أنواع من ناحية الخطورة حسب الجدول التالي :-
نسبه الحالات المصابة ( 40% من حالات الهيموفيليا ) مستوى عامل 8 أو 9 في الدم * ( أقل من 1% ) تصنيف الحالات ( شديدة )
نسبه الحالات المصابة ( 20% - 25% من حالات الهيموفيليا ) مستوى عامل 8 أو 9
في الدم * ( 1- 5% من النسبة الطبيعية) تصنيف الحالات ( متوسطة)
نسبه الحالات المصابة ( 35% - 40% من حالات الهيموفيليا ) مستوى عامل 8 أو 9
في الدم * ( 5- 30% من النسبة الطبيعية ) تصنيف الحالات ( بسيطة )
*نشاط عامل التخثر لدى الشخص الطبيعي يعتبر 100% ويتراوح ما بين 50% -150%
المرضى المصابون بالهيموفيليا الشديدة يكون لديهم مستوى عامل 8 أو عامل 9
أقل من 1% من المستوى الطبيعي في الدم ، وبدون علاج وقائي يمكن أن يصابوا
بالنزف عدة مرات في الشهر، وليس هناك سبب واضح للنزيف ، وهذا يدعى النزيف
التلقائي.
والمصابون بالهيموفيليا متوسطة الدرجة عادة ينزفون بدرجة أقل في معظم وهذا
النزف يتكرر بسبب الإصابة برضة غير خطيرة مثل جروح الرياضة ، ومع ذلك فبعض
المصابين بالهيموفيليا متوسطة الدرجة خصوصا أولئك الذين لديهم مستوى عامل 9
أو عامل 8 حوالي 2% أو أقل يمكن أن يتكرر عندهم نزف الدم العفوي وبنفس
الطريقة التي تحدث عند الأشخاص المصابين بمرض الهيموفيليا الشديدة.
أما الأشخاص المصابين بالهيموفيليا البسيطة فإنهم يصابون أيضا بالنزيف ولكن
بدرجة أقل بكثير، وربما يدركون وجود مشكلة النزيف فقط في حالة إجراء جراحة
أو خلع سن أو إصابة خطيرة. والخطر بالنسبة للمرضى المصابين بالهيموفيليا
البسيطة يتمثل في أنهم يصابون بحالات نزيف قليلة جدا وبالتالي فهم عادة لا
يعرفون ماذا يفعلون عندما يحدث النزيف ، والنساء الحاملات لهذا المرض ربما
ينزفن أكثر خلال الدورة الشهرية ، ولهذه الأسباب فالأشخاص المصابون
بالهيموفيليا البسيطة هم أيضا بحاجة لمتابعة حالتهم في مركز علاج
الهيموفيليا. لمزيد من المعلومات أنظر الهيموفيليا البسيطة والمتوسطة
هل هناك علاج فعال لمرض الهيموفيليا ؟
نعم ، ولله الحمد ، يوجد علاج فعال، والأدويه الحالية لهيموفيليا- أ و
هيموفيليا- ب فعالة جدا ، وأساس العلاج لهذا المرض هو استخدام عامل التخثر
المركز حيث يتضمن هذا العلاج إدخال عامل التخثر المفقود إلى الدم عند
الأطفال المصابين بهذا المرض، وهي طريقة آمنة وفعالة لإيقاف النزيف ، وحتى
هذا العلاج يمكن أن يستعمل كطريقة وقائية لمنع النزيف من الحدوث كليا.
وفي هذه الأيام فإن الأطفال الذين يولدون مصابين بهذا المرض يمكن أن تنتظرهم حياة صحية ونشطة كغيرهم من الاطفال .
المضاعفات يمكن أن تحدث فعلا، ومن أخطرها ظهور المثبطات فبعض المصابين
بالهيموفيليا لا يستجيب جهازهم المناعي بشكل فعال لعامل التخثر المركز الذي
يتم تزويدهم به لوقف أو منع النزيف نتيجه لتكون المثبطات ، فالعامل المركز
يتعرف عليه الجسم كمادة غريبة عليه ولأن دفاعات الجسم لا تميزه فإن أجهزة
المناعة في الجسم تقاوم هذا الجسم الغريب بتكوين أجسام مضادة وهي مواد
طبيعية تسبح في الدم ، والأجسام المضادة التي تكونت في الجسم تبطل مفعول
العامل المركز المحقون في الوريد ومن ثم تمنعه من القيام بعمله أي وقف
النزيف، وهذه الأجسام المضادة تسمى المثبطات ، ولحسن الحظ هناك علاجات
فعالة للأطفال الذين تتكون لديهم المثبطات.
كيف يتخثر الدم في الوضع الطبيعي؟
ينتقل الدم في أنحاء الجسم داخل شبكة من الأوعية الدموية، وعندما تجرح
الأنسجة يحدث ضرر في الأوعية الدموية يسبب تسرب الدم من خلال ثقوب في جدران
تلك الأوعية ، وقد تنفتح أو تجرح الأوعية القريبة من سطح الجلد كما في
حالة جروح الجلد أو قد تجرح في أماكن عميقة داخل الجسم ما ينتج عنه كدمة أو
نزيف داخلي ، لمزيد من المعلومات وللتعرف على أنواع النزيف المختلفة أنظر
التعامل مع حالات نزف الدم .
أن الجلطة أو التخثر هي عملية معقدة تجعل من الممكن إيقاف نزف الأوعية
الدموية المجروحة وحالما تجرح جدران الأوعية الدموية تعمل البروتينات معا
لتكوين جلطة تشكل سدادة للجرح وهناك عدة خطوات تساهم في تكوين هذه السدادة
في مكان الإصابة.
والمتأمل في الخطوات والمراحل التاليه التي يتخذها الجسم في التعامل مع النزيف خير دليل على أعجاز الخالق سبحانه وتعالى
المرحلة الاولى : تتقلص الأوعية الدموية لتبطئ تدفق الدم للمنطقة المجروحة. وهذا يدعى انقباض الأوعية .
المرحلة الثانيه : الصفائح الدموية ، وهي عبارة عن قطع صغيرة جدا من
الخلايا تكون أول من يصل إلى مكان الإصابة حيث تلعب الصفائح دورا هاما في
عملية وقف النزيف عن طريق تجمعها معا وبذلك يبدأ إصلاح وعلاج الأوعية
الدموية المجروحة. وهذا يدعى التصاق الصفائح.
المرحلة الثالثه : تبعث هذه الصفائح إشارات كيميائية تدعوا فيها الصفائح
الدموية الأخرى وعوامل التخثر لمساعدتها مثل عامل ويلبراند، وهذه الصفائح
المنتشرة تطلق المواد التي تنشط الصفائح المجاورة الأخرى والتي تتجمع معا
في مكان الجرح وتشكل سدادة من الصفائح، وهذه العملية تسمى تجمع اوتكدس
الصفائح .
المرحلة الرابعه : يشكل سطح هذه الصفائح المنشطة مكانا ملائما لحدوث الجلطة
حيث تتحد مع عوامل التخثر الموجوده في تيار الدم لتكوين سلسلة تسمى
الفيبرين حيث تنتظم حزم الفيبرين معا لنسج شبكة حول الصفائح الدموية وهذا
يمنع الصفائح الدموية من الانزلاق والعودة في تيار الدم. وهذه البروتينات
اوالعوامل 1، 2، 5، 7،8، 9، 10،11، 13 تعمل مثل قطع الدومينو في التفاعل
المسلسل وهذه العملية تسمى حاجز التخثر.
ما هي مشكلة التخثر في حالة الإصابة بمرض الهيموفيليا ؟
عندما يُفقَد أحد البروتينات (على سبيل المثال عامل 8 ) فإن سلسلة التفاعل
تنقطع ولا يحدث التخثر، أو يحدث بصورة أبطأ بكثير من الوضع الطبيعي.
والصفائح الدموية في مكان الجرح لا تتشابك في مكان الإصابة لتكوين جلطة
دموية دائمة ، وتكون الجلطة طرية حيث تسهل إزاحتها من مكانها ، وبدون علاج
يمكن أن يستمر النزيف لعدة أيام وأحيانا عدة أسابيع ويتكرر عادة حدوث
النزيف.
DREAM MAN- مدير موقع المعهد الطبي
- تاريخ التسجيل : 25/01/2010
عدد المساهمات : 533
المزاج : رايق
الجنس :
رد: امراض الدم الوراثية بالتفصيل و بالعربي
من طرف DREAM MAN الخميس يناير 27, 2011 11:52 pm
الثلاسيميــــــــا //


الانيميــــا المنجليـــــة //

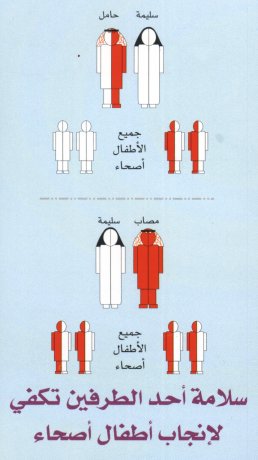
انيميــــــــا الفـــــول //



الهيموفيليــــــــــــا //



الانيميــــا المنجليـــــة //
انيميــــــــا الفـــــول //
الهيموفيليــــــــــــا //
DREAM MAN- مدير موقع المعهد الطبي
- تاريخ التسجيل : 25/01/2010
عدد المساهمات : 533
المزاج : رايق
الجنس :
» ابيضاض الدم النقوي المزمن
» جمع عينات الدم Collection of Blood
» الصورة التي يحلل بها الدم
» شرح لمادة علم الدم hematology
» صورة الدم كاملة CBC
» جمع عينات الدم Collection of Blood
» الصورة التي يحلل بها الدم
» شرح لمادة علم الدم hematology
» صورة الدم كاملة CBC
صفحة 1 من اصل 1
صلاحيات هذا المنتدى:
لاتستطيع الرد على المواضيع في هذا المنتدى